КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Кондуктометрия используется также для определения констант равновесия химических реакций в растворах, констант диссоциации слабых электролитов
Кондуктометрический метод анализа – один из наиболее точных способов определения растворимости труднорастворимых соединений. Он основан на измерении электропроводности жидкой фазы, находящейся в равновесии с соответствующим твердым соединением. Если известны подвижности ионов, на которые диссоциирует данное труднорастворимое соединение, то, определив удельную электропроводность раствора, можно вычислить его концентрацию.
Электроды сравнения. Электрод сравнения должен обладать постоянным потенциалом, не зависящим от состава исследуемого раствора. В качестве электродов сравнения чаще используют хлоридсеребряный и насыщенный каломельный электроды. Хлоридсеребряный электрод состоит из серебряной проволочки, электролитически покрытой слоем хлорида серебра и погруженной в раствор хлорида калия. Для полуреакции
AgClтв + e- → Ag0 + Cl-
зависимость потенциала электрода от активности хлорид-ионов описывается уравнением
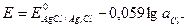
Иногда электроды второго рода используют в качестве индикаторных, с их помощью измеряют концентрацию ионов, не участвующих непосредственно в процессе переноса электрона.
Прямая потенциометрия основана на непосредственном измерении потенциала индикаторного электрода и вычислении активности потенциалопределяющих ионов по уравнению Нернста.
Метод широко применяется для определения концентрации водородных ионов или рН растворов. Создание надежно работающих ионоселективных электродов значительно расширило практические возможности прямого метода. Прямой потенциометрический метод часто стали называть ионометрическим методом анализа или ионометрией.
Это удобный, простой и экспрессный современный метод: продолжительность анализа определяется временем подготовки пробы, поскольку, непосредственно на измерение тратится не более 1–2 мин.
В методе ионометрии предварительно, пользуясь растворами с известной концентрацией, градуируют электрод, т.е. опытным путем определяют зависимость его потенциала от концентрации потенциал-определяющего иона. Затем измеряют потенциал раствора с неизвестной концентрацией определяемого иона и по градуировочному графику находят его содержание.
Ионоселективные электроды позволяют измерять концентрации ионов до 10‾6 М в растворе. При этом необходимый для определения объем раствора составляет всего 0.05–0.1 мл.
Потенциометрическое титрование основано на определении точки эквивалентности по изменению потенциала индикаторного электрода при проведении химической реакции между титрантом и определяемым веществом. Вблизи точки эквивалентности происходит резкое изменение (скачок) потенциала индикаторного электрода, если хотя бы один из участников реакции титрования является участником электродного процесса.
Кривые титрования могут быть построены в координатах: потенциал индикаторного электрода (Е) ‑ объем титранта (V) (рис. 2а.). Это так называемая интегральная кривая потенциометрического титрования. Точка перегиба на кривой отвечает точке эквивалентности. Ее находят графическим путем: нахождением середины отрезка между касательными двух ветвей кривой.
Для более точного нахождения точки эквивалентности часто строят дифференциальную кривую потенциометрического титрования в координатах ∆Е / ∆V ‑ V (рис. 2б). На точку эквивалентности указывает максимум полученной кривой, а отсчет по оси абсцисс, соответствующий этому максимуму, дает объем титранта, израсходованного на титрование до точки эквивалентности.
На рис. 2в представлена кривая потенциометрического титрования в координатах: вторая производная потенциала по объему титранта ∆ 2 Е / ∆ 2 V ‑ объем титранта, V. Для нахождения точки эквивалентности соединяют концы обеих ветвей кривой.
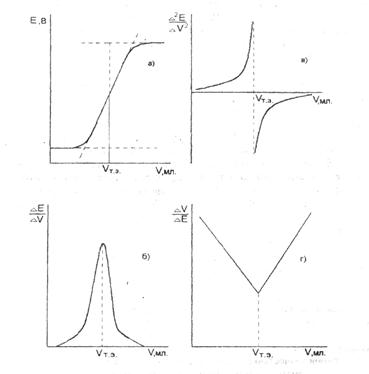
Рис. 2.Кривые потенциометрического титрования. а) интегральная кривая; б) дифференциальная кривая; в) кривая титрования по второй производной; г) кривая Грана.
В методе Грана (рис. 2г) точка эквивалентности определяется по графику в координатах: ∆V / ∆E ‑ V. Перед точкой эквивалентности и после нее кривая Грана линейна. Точка эквивалентности находится как точка пересечения этих прямых. Достоинства и удобства метода Грана особенно заметны при анализе разбавленных растворов, позволяющих определить точку эквивалентности с достаточной точностью вследствие линейности графика, а также в тех случаях, когда кривая титрования выражена плохо.
В потенциометрическом титровании могут быть использованы любые известные типы химических реакций, протекающие быстро и количественно.
Кислотно-основное потенциометрическое титрование основано на протекании химической реакции нейтрализации. В качестве индикаторного применим любой электрод с водородной функцией: водородный, хингидронный, стеклянный. Чаще всего используется стеклянный электрод. Метод позволяет провести количественное определение компонентов в смеси кислот, если константы их диссоциации различаются не менее чем на три порядка (например, в смеси соляной и уксусной кислот); многоосновных кислот (оснований), так как удается достичь разделения конечных точек многоступенчатого титрования (на кривой титрования при этом наблюдается несколько скачков).
Широкие возможности анализа многокомпонентных смесей без разделения открывает применение неводных растворителей. Например, раздельное определение соляной и монохлоруксусной кислот невозможно в водном растворе из-за отсутствия двух скачков титрования, но его удается провести в ацетоне.
В окислительно-восстановительном потенциометрическом титровании наибольшее распространение нашелплатиновый индикаторный электрод. Величина скачка определяется разностью формальных потенциалов полуреакций. Желательно, чтобы одна из полуреакций была обратимой. При титровании не рекомендуется измерять потенциал до добавления титранта и вблизи точки эквивалентности, т.к. приобретаемый электродом смешанный потенциал неустойчив, поэтому его трудно измерить.
Комплексонометрическое потенциометрическое титрование используется для определения катионов металлов при титровании их комплексоном (III) (ЭДТА) с применением в качестве индикаторного соответствующего металлического электрода: титрование солей меди с медным электродом, солей цинка ‑ с цинковым электродом и т.д., а также ртутного электрода. Также используют ионоселективные электроды, обратимые относительно определяемого компонента. В ряде случаев необходимо добавление в анализируемый раствор потенциометрических индикаторов – потенциалопределяющих ионов, вводимых в небольшом количестве и обеспечивающих отклик индикаторного электрода либо до, либо после достижения конечной точки титрования (так, при титровании железа (Ш) вводят железа(П) в небольшом количестве).
В осадительном потенциометрическом титровании индикаторными электродами служат металлические или мембранные электроды, чувствительные к определяемому иону или иону - осадителю.
Например, можно определять галогенид-ионы (Сl‾, Вr‾, I‾) на серебряном электроде титрованием нитратом серебра. До точки эквивалентности потенциал электрода зависит от активности галогенид-ионов и серебряный электрод является электродом II рода. За точкой эквивалентности при избытке ионов серебра потенциал электрода зависит от активности собственных ионов (электрод I рода). Величина скачка зависит от растворимости осадка. Можно провести дифференцированное титрование смеси хлорид-, бромид- и иодид-ионов.
По методу осаждения могут быть также определены катионы серебра, ртути, цинка, свинца и т. д.
Существует несколько вариантов потенциометрического титрования в зависимости от инструментальных особенностей. С применением неполяризованных электродов можно провести титрование а) с одним индикаторным электродом и одним электродом сравнения; б) с двумя различными индикаторными электродами. Варианты титрования с применением поляризованных электродов (титрование под током): а)с одним индикаторным электродом и одним электродом сравнения; б) с двумя одинаковыми электродами сравнения.
Метод потенциометрического титрования имеет ряд преимуществ перед прямой потенциометрией и титрованием с визуальными индикаторами: отсутствие искажения результатов за счет диффузионного потенциала; нет необходимости знать коэффициент активности определяемого иона; исключение субъективных ошибок за счет инструментального фиксирования конечной точки; возможность анализа мутных и окрашенных растворов; сравнительно легкая автоматизация; возможность дифференцированного титрования компонентов смеси, в том числе с использованием неводных растворителей. Результаты определений методом потенциометрического титрования более точны, чем при использовании прямой потенциометрии, так как вблизи точки эквивалентности небольшому изменению концентрации соответствует большое изменение потенциала индикаторного электрода.
К недостаткам потенциометрического титрования можно отнести не всегда быстрое установление потенциала после добавления титранта.
3. Кулонометрические методы основаны на измерении количества электричества, затраченного на электропревращение определяемого вещества (прямая кулонометрия) или на получение промежуточного реагента, который количественно реагирует с определяемым веществом (косвенная кулонометрия).
В основе кулонометрических методов анализа лежат законы электролиза Фарадея:
1. Количество (масса) вещества, выделившегося при электролизе, пропорциональна количеству электричества, прошедшего через раствор.
2. При прохождении через раствор одного и того же количества электричества, на электродах выделяется одно и то же количество эквивалента вещества.
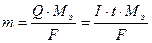
где m – масса вещества, выделившегося при электролизе, г;
Q ‑ количество электричества, Кл;
Мэ ‑ молярная масса эквивалента, г/моль-экв;
F ‑ число Фарадея: F = 96500 Кл/моль-экв;
I ‑сила тока, А;
t ‑ время электролиза, с.
Обязательным является условие, что электропревращение вещества на электроде происходит со 100%-ной эффективностью, т.е. со 100%-ным выходом по току, что возможно только в отсутствие побочных процессов (разложение воды, окисление или восстановление примесей, участие материала электрода в электрохимической реакции и др.)
Электролиз в кулонометрической ячейке можно проводить либо при постоянной силе тока (гальваностатическая кулонометрия), либо при постоянном потенциале (потенциостатическая кулонометрия).
Прямая кулонометрия. Метод прямой кулонометрии пригоден для определения только электроактивных веществ, поскольку, в его основе лежит непосредственное электропревращение вещества на электроде. Прямые кулонометрические измерения можно проводить, поддерживая постоянной либо силу тока (необходимо иметь гальваностат), либо потенциал рабочего электрода (необходимо иметь потенциостат).
Если электролиз проводят при постоянной силе тока (гальваностатическая кулонометрия), то количество электричества (Q) за время электролиза tЭ, при постоянном токе I равно:
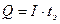
Погрешность измерения Q зависит от точности измерения времени, поскольку современные приборы позволяют очень точно измерять даже небольшие токи. Прямая кулонометрия при постоянной силе тока является более простым, но менее селективным способом, поскольку в определенный момент времени может пойти реакция с участием мешающих веществ, фонового электролита или растворителя, и выход по току начинает уменьшаться по экспоненциальному закону.
Чаще применяют прямую кулонометрию при постоянном потенциале рабочего электрода. Потенциал электрода выбирают в области предельного тока; в этом случае ток, протекающий через ячейку, будет уменьшаться по экспоненциальному закону в соответствии с уменьшением концентрации электроактивного вещества (рис. 3.).
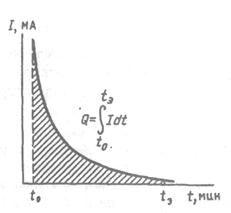
Рис. 3. Определение количества электричества в методе прямой кулонометрии
Можно самописцем записать изменение силы тока как функцию времени и найти количество электричества, измерив площадь под кривой планиметром (графическое интегрирование), однако, этот простой способ не очень точен и не годится для количественного анализа.
Можно использовать химические интеграторы (кулонометры). Кулонометр – это электролитическая ячейка, в которой при замыкании цепи со 100%-ным выходом по току протекает электрохимическая реакция известной стехиометрии. Кулонометр включают последовательно с кулонометрической ячейкой, поэтому за время электролиза через обе ячейки протекает одинаковое количество электричества. По окончании электролиза по массе выделенного в кулонометре вещества рассчитывают эквивалентное ему количество электричества:
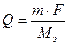
Однако в аналитической практике этот способ измерения Q применяют редко. Чаще измеряют ток, а не количество электричества. Величина тока в любой момент времени определяется формулой:

где It и I0 – сила тока в момент времени t и в начальный момент электролиза соответственно; k =0.43 SD / Vδ – коэффициент, зависящий от природы электроактивного вещества и от условий электролиза (S – площадь поверхности электрода, D – коэффициент диффузии вещества, V – объем раствора, δ – толщина диффузионного слоя).
Электролиз ведут до достижения остаточного тока It, величина которого определяется требуемой точностью. Так, если допустима погрешность порядка 0.1%, то электролиз можно считать завершенным при It ~ 0.001· I0.
Прямая кулонометрия – высокочувствительный и точный метод анализа, легко поддающийся автоматизации. Общая погрешность метода может составлять 0.5%. При проведении электролиза в течение 103 с при силе тока 1 мкА принципиально возможно определить до 10‾9 г вещества.
Кулонометрическое титрование обычно проводят, поддерживая постоянной силу тока. Этот метод применяется для определения и электроактивных и электронеактивных веществ. В процессе титрования определяемое вещество реагирует с титрантом, образующимся в результате электрохимической реакции на электроде. Такой титрант называют электрогенерированным кулонометрическим титрантом, а электрод, на котором его получают – генераторным. Вторым электродом схемы генерации является так называемый вспомогательный электрод. Его обычно изолируют от анализируемого раствора, помещая в трубку с дном из пористого стекла, так как продукт реакции на вспомогательном электроде нередко мешает кулонометрическому определению. Индикаторными электродами могут быть два платиновых или золотых электрода, если для индикации применяется амперометрический метод, или платиновый и каломельный или хлоридсеребряный, если используется потенциометрическая индикация.
Электрогенерированный титрант можно получать из воды (ОН‾ при восстановлении ее на катоде или Н+ при окислении на аноде), растворов солей, кислот, вспомогательных реагентов (например, при окислении KI можно получить I2), твердых электроактивных рабочих электродов.
Электрогенерированный титрант можно получать непосредственно в ячейке для кулонометрического титрования (внутренняя генерация) или в отдельном устройстве (внешняя генерация), а затем вводить его в кулонометрическую ячейку. Для обеспечения 100%-ной эффективности тока необходимо ввести избыток вспомогательного реагента (это реализуется при генерации титранта из воды или материала электрода). В этом случае протекание конкурирующих реакций на электроде исключается, и по количеству электричества, затраченного на генерацию титранта, можно будет правильно рассчитать содержание определяемого вещества. Примеры электрогенерированных кулонометрических титрантов приведены в табл. 2.
В качестве химической реакции между кулонометрическим титрантом и определяемым веществом может быть использована любая химическая реакция, применяемая в титриметрии – реакции кислотно-основного взаимодействия, окисления-восстановления, осаждения, комплексообразования.
Для определения конца кулонометрического титрования пригодны практически все способы установления конечной точки в титриметрии:
использование визуальных индикаторов (крахмала, фенолфталеина) и инструментальных методов. Наибольшее распространение получили потенциометрический и амперометрический методы с двумя индикаторными электродами.
К числу достоинств кулонометрического титрования следует отнести то, что нет необходимости в приготовлении, стандартизации и хранении титранта, т.к. он образуется в процессе титрования и сразу же расходуется. При электрогенерации можно получать титранты, крайне неустойчивые в обычных условиях хранения, например, стандартные растворы Cu(1), Cr(П), Ag(Ш), или легколетучие вещества – Cl2, Br2. Регулируя силу тока, можно прибавлять титрант сколь угодно малыми порциями, что более удобно, чем при использовании обычной бюретки. Метод кулонометрического титрования характеризуется высокой чувствительностью и точностью (0,05-0,1%), позволяя прямым титрованием определять вещества в растворе при концентрации до 10-6 моль/л, что намного превышает возможности других титриметрических методов. Метод легко автоматизируется.
4. Вольтамперометрический метод анализа основан на изучении поляризационных или вольтамперных кривых (вольтамперограмм) – зависимостей силы тока от приложенного напряжения. Вольтамперограммы регистрируют в электролитической ячейке с помощью поляризуемого индикаторного электрода и неполяризуемого электрода сравнения, погруженных в анализируемый раствор. На легкополяризуемом микроэлектроде происходит электровосстановление или электроокисление вещества (деполяризатора).
В настоящее время существует несколько десятков разновидностей вольтамперометрии, способных обеспечить экспрессность, высокую чувствительность, избирательность при определении неорганических и органических веществ в самых разнообразных объектах.
В классическом полярографическом методе в качестве рабочего электрода используют ртутный капающий электрод (ртуть вытекает из тонкого капилляра), электродом сравнения служит насыщенный каломельный электрод или донная ртуть. Если в растворе присутствуют вещества, способные электрохимически восстанавливаться или окисляться (так называемые деполяризаторы), то при наложении на электрохимическую ячейку линейно-меняющегося потенциала регистрируется вольтамперная кривая в виде волны (рис. 4).

Рис. 4. Классическая полярограмма: 1 – остаточный ток, 2 – диффузионный ток
При низких значениях потенциала (участок 1), величина которого не достаточна для того, чтобы на рабочем микроэлектроде проходила электрохимическая реакция, через ячейку проходит очень незначительный остаточный ток. Остаточный ток обусловлен прежде всего током заряжения двойного электрического слоя, который образуют ионы раствора на катоде, когда потенциал электрода недостаточен для их разряда, и присутствием в растворе более электрохимически активных, чем определяемое вещество, примесей.
При увеличении потенциала электрохимически активное вещество – деполяризатор вступает в электрохимическую реакцию на электроде, например,
Cd2+ + 2 е + Hg  Cd (Hg)
Cd (Hg)
и в результате этого ток резко возрастает. Это так называемый фарадеевский ток. С ростом потенциала ток возрастает до некоторого предельного значения, оставаясь затем постоянным (участок 2). Предельный ток обусловлен тем, что в данной области потенциалов практически весь деполяризатор из приэлектродного слоя исчерпан в результате электрохимической реакции, а обедненный слой обогащается за счет диффузии деполяризатора из объема раствора. Скорость диффузии деполяризатора в этих условиях контролирует скорость электрохимического процесса в целом, и ток перестает зависеть от наложенного напряжения. Такой ток называют предельным диффузионным.
Для того, чтобы исключить электростатическое перемещение деполяризатора (миграцию) в поле электродов и понизить сопротивление ячейки, измерения проводят в присутствии большого избытка сильного электролита, называемого фоновым или фоном. Являясь электрохимически индифферентным, он не принимает участия в электродной реакции, но его ионы экранируют электрод, уменьшая тем самым движущую силу миграции под действием электрического поля практически до нуля.
Полярограмма содержит ценную аналитическую информацию: качественной характеристикой деполяризатора является потенциал полуволны (Е 1/2 ) – потенциал, при котором ток равен половине величины диффузионного тока. Потенциал полуволны Е 1/2 не зависит от силы тока и концентрации восстанавливающегося иона, зависит от его природы. Определение Е 1/2составляет основу качественного полярографического анализа.
Предельный диффузионный ток (I d)линейно связансконцентрацией деполяризатора в объеме раствора, и эта зависимость является основой количественного полярографического анализа. Связь I d с концентрацией иона С м выражается уравнением Ильковича:
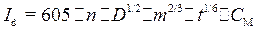
где: п – заряд иона; D – коэффициент диффузии, см2·сˉ1; т – скорость вытекания ртути, мг·сˉ1; t – время образования капли (период капания), с; CM – концентрация деполяризатора, ммоль/л; I d – ток, мкА.
Если в растворе находится несколько электрохимически активных соединений, на полярограмме будет не одна волна, а несколько ‑ по числу восстанавливающихся ионов (рис. 5). Можно получить полярографический спектр ионов и затем по измеренному Е 1/2идентифицировать неизвестное вещество.
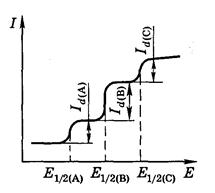
Рис. 5. Полярограмма при наличии в растворе восстанавливающихся веществ А, В и С
Для определения концентрации используют метод сравнения со стандартом, метод градуировочного графика и метод добавок.
Амперометрическое титрование. Полярографический метод можно применить для определения точки эквивалентности в титриметрических методах анализа, если хотя бы один из участников реакции или ее продукт электроактивны – окисляются или восстанавливаются на микроэлектроде. Это так называемый метод амперометрического титрования. Титрование проводят при заданном значении потенциала, соответствующем достижению предельного диффузионного тока. Связь между вольтамперными кривыми и кривой зависимости предельного тока от объема титранта представлена на рис. 6.
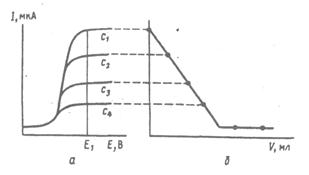
Рис. 6. Вольтамперограммы электроактивного вещества при концентрациях с1>c2>c3>c4 (а), кривая амперометрического титрования этого вещества при потенциале индикаторного электрода E 1 (б)
В ходе амперометрического титрования регистрируют величину диффузионного тока в зависимости от объема добавленного титранта. Кривая амперометрического титрования в координатах: сила тока ‑ объем титранта (I d ‑ V) состоит из двух линейных участков, точку эквивалентности находят графически. В качестве индикаторных электродов в амперометрическом титровании обычно применяют платиновые, графитовые и другие твердые электроды, чаще всего вращающиеся.
Следует различать электрохимическую реакцию, протекающую на границе раздела фаз электрод-раствор, и химическую реакцию, протекающую в растворе между определяемым веществом и титрантом.
Вид кривой амперометрического титрования зависит от того, какой компонент химической реакции участвует в электродном процессе (является деполяризатором): определяемое вещество, титрант или продукт реакции.
В методах амперометрического титрования используют реакции осаждения, комплексообразования и окисления ‑ восстановления. Многие анионы: Сl‾, Вr‾, I‾, SO42‾, МоO42‾ и др. титруются солью свинца, при этом регистрируется ток восстановления Рb2+ на ртутном капающем электроде.
В реакциях осаждения часто применяется осаждение органическими реагентами: 8-оксихинолином, купфероном, диметилглиоксимом и др., причем титрование можно проводить как по току восстановления катиона, так и по току органического реагента.
Широко используется в амперометрическом титровании реакция образования этилендиаминтетраацетатных комплексов с различными катионами: Bi3+, Fe3+, Fe2+, Ni2+, Pb2+, Zn2+, Cu2+, Co2+, Cd2+.
При амперометрическом титровании с использованием реакций окисления – восстановления в качестве титрантов используют К2Сr2О7; Ce(SO4)2; КBrO3 и I2 для определения восстановителей; FeSO4, Na2S2O3 – для определения окислителей.
Титрование с двумя индикаторными электродами. В анализируемый раствор погружают два одинаковых инертных электрода, например, платиновых, между которыми с помощью внешнего источника поддерживается небольшая разность потенциалов (10–50 мВ) и в ходе титрования отмечают силу тока. До начала титрования ток практически равен нулю, так как в отсутствие окислительно-восстановительной пары при столь малой разности потенциалов электродные процессы не происходят. После введения титранта в растворе появляются две окислительно-восстановительные пары. Чем больше обратимость редокс-системы, тем меньшее напряжение требуется налагать на электроды. Возникновение тока в ячейке связано с протеканием электрохимических процессов на обоих электродах. Вид кривых титрования зависит от обратимости катодного и анодного процессов. Для полностью обратимой пары определяемого вещества (например, Fe3+/Fe2+), окисленная форма которого восстанавливается на катоде:
Fe3+ + e → Fe2+,
а восстановленная форма окисляется на аноде:
Fe2+ - e → Fe3+,
максимум тока будет наблюдаться при равенстве концентраций окисленной и восстановленной форм, когда раствор оттитрован на 50%. Поскольку индикаторные электроды одинаковы, одинаков и вклад катодного и анодного процессов в величину тока – кривая титрования симметрична, до начала титрования и в точке эквивалентности ток равен нулю. Если окислительно-восстановительная пара титранта необратима, ток после точки эквивалентности остается равным нулю (рис. 7 б), если пара титранта обратима, то после точки эквивалентности ток возрастает за счет участия в электродном процессе пары титранта (рис. 7. в).
Примером реакции, в которой обратимая система титруется необрати мой, является перманганатометрическое определение соли Мора:
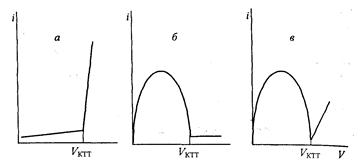
Рис. 7. Кривые амперометрического титрования с двумя поляризованными электродами: а – титрование необратимой редокс-системы электрохимически обратимым титрантом; б – титрование обратимой редокс-системы электрохимически необратимым титрантом; в – титрование обратимой редокс-системы электрохимически обратимым титрантом
5Fe2+ +MnO4 - + 8H+ → 5Fe3+ + Mn2+ +4H2O
В реакции титрования железа (П) солью церия (1V):
Fe2+ + Ce4+ → Fe3+ + Ce3+
обе окислительно-восстановительные пары обратимы.
В методе биамперометрического титрования часто отпадает необходимость в построении кривой титрования, т.к. точка эквивалентности может быть определена по резкому прекращению или появлению тока.
Достоинством метода амперометрического титрования являются его экспрессность и простота, этим методом можно определять практически все элементы периодической системы и большое число органических соединений, причем определяемое вещество может не проявлять электрохимической активности. Основным достоинством метода является возможность анализа многокомпонентной смеси без предварительного разделения, достаточно высокая точность и чувствительность. Воспроизводимость результатов лучше, чем в полярографическом методе, поскольку регистрируют изменение тока в ходе титрования, и отпадает необходимость удалять из раствора кислород.
5. Кондуктометрия - метод физико-химического анализа, основанный на измерении электропроводности растворов. Она обладает рядом преимуществ перед химическими методами анализа, так как позволяет определять содержание индивидуального вещества в растворе простым измерением электропроводности раствора. Для этого нужно только иметь предварительно вычерченную калибровочную кривую зависимости электропроводности от концентрации вещества. Далее, в процессе измерения электропроводности анализируемый раствор практически не изменяется, благодаря чему можно проводить повторные измерения и, сохранив его, в любое время проверить полученные результаты.
|
|
Дата добавления: 2014-01-20; Просмотров: 1447; Нарушение авторских прав?; Мы поможем в написании вашей работы!