КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Випаровування металів і сплавів
|
|
|
|
Утворення вакуумних покриттів визначається послідовним проходженням складних фізико-хімічних процесів, а саме:
• випаровуванням чи розпиленням вихідного матеріалу покриття;
• напрямленим масоперенесенням у вигляді потоку атомів чи іонів матеріалу покриття на поверхню основи;
• співударом потоку з поверхнею і подальшою адсорбцією чи десорбцією атомів чи іонів на ній;
• поверхневою дифузією атомів до місць найкращого утворення зародків покриття;
• міграцією і коалесценцією зародків та утворенням острівців; зрощенням острівців у суцільну плівку; ростом суцільної плівки і утворенням покриття необхідної товщини.
Вказані процеси залежать від ступеня вакууму в камері напилення. Цей показник визначається співвідношенням між відстанню L від випарника до поверхні і середньою довжиною вільного пробігу атомів l. Якщо l < L, то такий вакуум називають низьким, якщо l = L – середнім і якщо l > L – високим.
У низькому вакуумі траєкторія руху кожного атома має вигляд ламаної лінії внаслідок багатократного зіткнення з молекулами довколишнього газу. Потік пари перемішується, що дає змогу наносити в такому паровому середовищі покриття, рівномірні за товщиною.
У високому вакуумі рух атомів здійснюється по прямих лініях, незалежно один від одного і без зіткнень до конденсації і утворення покриття.
Форма і товщина покриття визначаються формою і густиною потоку випаровуваних атомів.
У середньому вакуумі спостерігаються явища, характерні для низького і високого вакуумів.
Згідно з кінетичною теорією газів, середня довжина вільного пробігу молекул прямо пропорційна температурі (Т) й обернено пропорційна тиску (Р) газу:
|
|
|
 (1)
(1)
де k – стала Больцмана; d – ефективний діаметр молекули (за припущенням, що вони є пружними кульками).
При тиску р = 133×10-1 Па середня довжина вільного пробігу молекул становить приблизно 500 см, і рух атомів можна вважати прямолінійним. Подальше зниження тиску збільшує довжину вільного пробігу до десятків метрів, і випаровування відбувається у вигляді молекулярних потоків без зіткнення і розсіяння атомів.
У високому вакуумі швидкість випаровування атомів визначається співвідношенням Ленгмюра:
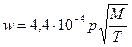 (2)
(2)
де р – тиск насиченої пари, Па; М – молекулярна маса речовини; Т – температура випаровування, К.
Температурою випаровування умовно вважають таку, при якій тиск насиченої пари матеріалу, що випаровується, становить 1,33 Па.
Залежність тиску (чи пружності) насиченої пари від температури визначається рівнянням Клапейрона-Клаузіуса, яке після розподілу змінних і інтегрування набуває вигляду:
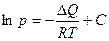 (3)
(3)
де DQ – теплота пароутворення.
Таким чином, нагрівання при випаровуванні різко збільшує тиск насиченої пари. Наприклад, підвищення температури на 10 – 15 % при випаровуванні Сr і Аl приводить до зростання тиску насиченої пари на порядок.
Пружність пари практично не залежить від тиску довколишнього газу. Тому термодинамічний вплив вакууму на процес випаровування незначний, і його можна не брати до уваги.
Вакуум суттєво впливає на кінетику процесу. Підвищення тиску залишкових газів у вакуумній камері вище, ніж 1,33×10-1 Па і підвищення температури поверхні матеріалу більше за значення, яке відповідає пружності пари 133 Па, різко підвищує імовірність зіткнення атомів між собою і з молекулами залишкових газів. Внаслідок цього відбувається зниження швидкостей випаровування порівняно з розрахунковими значеннями з рівняння Ленгмюра.
При випаровуванні сплавів тиск пари кожного компонента відрізняється від тиску над чистим металом і називається парціальним тиском даного компонента.
|
|
|
Для оцінки парціального тиску над рідким сплавом можна застосовувати закон Рауля. Наприклад, для бінарного сплаву компонентів А і В можна записати:
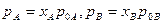 (4)
(4)
Де р А, р В – парціальні тиски пари компонентів рідкого сплаву; р 0А, р 0В – тиски пари чистих компонентів при температурі рідкого сплаву; х А, х В – молярні частки компонентів у сплаві х а + х в=1.
Якщо р 0А» р 0В, то
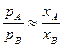 (5)
(5)
і проходить узгоджене випаровування, тобто склад сплаву і склад пари у процесі випаровування практично незмінний.
Для більшості реальних сплавів спостерігається відхилення від закону Рауля. Для оцінки їх поведінки вводять емпіричну величину, яку називають активністю:
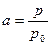 (6)
(6)
де р – тиск даного компонента над сплавом; p 0 – його тиск над чистою речовиною.
Активність пов'язана з молярною часткою через коефіцієнт активності f:
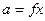 (7)
(7)
Залежно від типу реального сплаву коефіцієнт активності може бути більшим чи меншим від одиниці. Із врахуванням f парціальні тиски пари компонентів реального бінарного сплаву становлять:
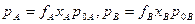 (8)
(8)
Щоб визначити швидкість випаровування кожного компоненту бінарного сплаву потрібно у формулу (2) підставити значення р А і р В із формули (8).
Якщо припустити, що коефіцієнт активності не залежить від концентрації, можна виділити емпіричний параметр, який характеризує бінарну систему:
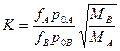 (9)
(9)
Найбільш досконалим способом нагрівання і випаровування матеріалу є електронно-променевий. Після проходження крізь електричне поле з різницею потенціалів U електрон прискорюється і набуває кінетичної енергії:
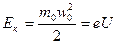 (10)
(10)
де m 0, w 0, е – маса, швидкість і заряд електрона відповідно.
Електрон зі стану спокою, пройшовши крізь поле з різницею потенціалів 1 В, матиме швидкість 593 км/с.
При зіткненні з поверхнею матеріалу кінетична енергія електрона витрачається на збудження рентгенівського випромінювання, утворення вторинних електронів і нагрівання. Основна частина кінетичної енергії перетворюється на теплову в тонкому поверхневому шарі. При прискорювальній напрузі 15 – 20 кВ глибина проникнення електрона в металеві матеріали становить 1 – 2 мкм. Основне гальмування електрона і виділення енергії відбувається в кінці пробігу, тобто температура сягає максимуму на певній відстані від поверхні.
|
|
|
Отже, при нагріванні електронним променем джерело теплоти знаходиться в самому тілі, в тонкому поверхневому шарі. Тиск електронного променя і конвекція, яка викликана градієнтом температури, забезпечують інтенсивне перемішування рідкої ванни. Тому можна вважати, що в рідкій ванні градієнт концентрації відсутній.
Для отримання однорідного покриття сплави з суттєво різним парціальним тиском компонентів випаровують з окремих незалежних джерел (рис. 20).
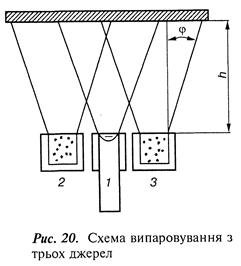
Випаровування таких сполук, як оксиди, карбіди, бориди, силіциди, нітриди, супроводжується зміною типу вихідних молекул. Для більшості тугоплавких сполук характерна дисоціація вихідних молекул з утворенням газоподібних продуктів.
Досвід показав, що практично без змін складу можуть бути отримані покриття сполук:
• оксиди – А12О3, Y2O3, MgO, ZrO2, SiO2;
• карбіди – TiC, ZrC, NbC;
• бориди – TiB2, ZrB2.
Інші сполуки, наприклад WC, SiC, AlN, TiN, ZrN, при нагріванні розпадаються з утворенням продуктів із різко відмінною леткістю і тому не можуть осаджуватись прямим випаровуванням.
Лекція 16. Створення атомарних, молекулярних та іонних потоків розпиленням твердого матеріалу без його переходу в рідкій стан. Реактивне випаровування і розпилення.
Створення атомарних, молекулярних та іонних потоків при нанесенні вакуумних покриттів здійснюється також розпиленням твердого матеріалу без його переведення в рідкий стан. Для розпилення застосовують іонне бомбардування, оскільки іони легко довести до необхідної швидкості.
Бомбардувальний іон вибиває атоми з вузлів кристалічної ґратки внаслідок передачі імпульсу. Атоми, розміщені на поверхні матеріалу, одразу переходять у газоподібний стан, а атоми, які знаходяться на деякій відстані від поверхні, передають імпульс іншим атомам ґратки, викликаючи каскад зіткнень (рис. 21).
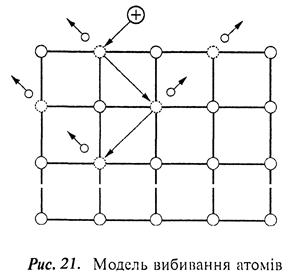
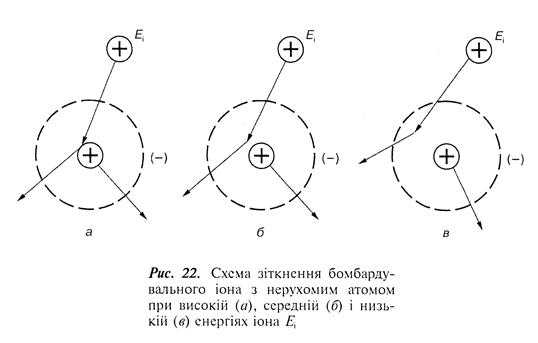
Особливості зіткнення падаючого іона залежать від його енергії. Енергії іонів поділяють таким чином (рис. 22): високі – коли іон пролітає поблизу ядра, і взаємодія зводиться до кулонівського відштовхування; проміжні – коли виявляється екранувальна дія електронних оболонок; малі – коли відбувається незначне проникнення в електронні оболонки.
|
|
|
Для початку процесу розпилення достатньо певного мінімального значення енергії іона, яка має назву порогової. Для більшості матеріалів порогова енергія розпилення змінюється від 10 до 100 еВ і залежить від типу бомбардувального іона, енергії зв'язку атомів у ґратці розпилюваного матеріалу. Зростання енергії іонів вище за порогове значення приводить до прискорення розпилення.
Інтенсивність розпилення характеризується коефіцієнтом розпилення як відношення кількості розпилених атомів N a до кількості бомбардувальних іонів N i:
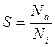 (12)
(12)
чи кількістю атомів речовини, вибитих одним іоном.
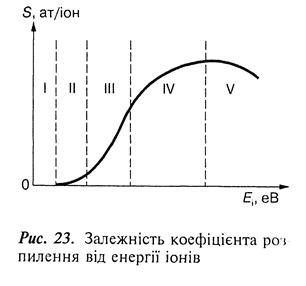
Залежність коефіцієнта розпилення від енергії іонів має кілька зон (рис. 23).
Зона І відповідає допороговим значенням енергії іонів і не фіксується сучасними методами вимірювань. У зоні II розпилєння починається з дуже малою швидкістю, але коефіцієнт розпилення різко зростає при невеликому підвищенні енергії іонів. У зоні III коефіцієнт розпилення збільшується приблизно лінійно з підвищенням енергії іонів. У зоні IV він продовжує збільшуватися з підвищенням енергії іонів, але з меншою швидкістю порівняно із зоною III. Це пояснюється більш глибоким проникненням іонів у матеріал, перешкоджає виходу вибитих атомів, оскільки вони вибиваються з глибоких шарів. Зона V характеризується пологим максимумом, у межах якого коефіцієнт розпилення практично не змінюється, а при подальшому підвищенні енергії іонів – починає зменшуватися внаслідок збільшення глибини проникнення бомбардувальних іонів.
При фіксованій енергії бомбардувального іона коефіцієнт розпилення залежить від атомного номера і структури електронних оболонок розпилюваного матеріалу. Так, в міру заповнення d -оболонок атомів розпилюваного матеріалу коефіцієнт розпилення збільшується. Найбільші значення коефіцієнта розпилення мають атоми із заповненими d -оболонками, наприклад Cu, Ag, Cd, Au. Аналогічна залежність спостерігається і для бомбардувальних іонів: максимальне розпилення викликають іони елементів із заповненими d -оболонками (Cu, Ag, Cd, Bi, Pd, Тl, Hg, Pt та ін.) і p -оболонками (інертні гази). Максимальний коефіцієнт розпилення 84 ат/іон встановлено при розпиленні Cd іонами Cd+ (саморозпилення з енергією 45 еВ).
Коефіцієнт розпилення залежить від енергії, маси і кута падіння іонів. Основними параметрами, що характеризують зіткнення іонів з кристалічною ґраткою, є переріз зіткнення іона з атомами кристалічної ґратки sг, при якому атому передається енергія, вища, ніж необхідна для зміщення атома з його нормального положення в ґратці Eа, і середня енергія Е, яка передається зміщеному атому.
Конденсатам, отриманим розпиленням, як правило, властиві більш досконала структура і висока адгезія до основи. Розпилення дає можливість отримувати багатокомпонентні системи без зміни хімічного складу конденсатів. Однаковий хімічний склад розпилюваної речовини і конденсату залишається також і в системах, компоненти яких мають коефіцієнти розпилення, що суттєво розрізняються. Це пояснюється структурно-кінетичними закономірностями розпилення багатокомпонентних систем. У перший момент розпилення з поверхні складного сплаву починає відділятися компонент, коефіцієнт розпилення якого найбільший. На поверхні формується так званий змінений шар, збіднений на компонент із великим коефіцієнтом розпилення. Швидкість розпилення цього компонента уповільнюється, в той час як відносна швидкість розпилення другого зростає. Під час подальшого бомбардування процес стабілізується: розпилення сплаву супроводжується збереженням складу і товщини зміненого шару, який виконує роль автоматичного регулятора швидкостей розпилення. Відмінність у складі виникає, коли катод, який розпилюється, має досить високу температуру, близьку до температури плавлення. При цьому конденсат збагачується на компонент із найбільшим коефіцієнтом розпилення.
Реакційне випаровування і розпилення. Деякі покриття отримують за допомогою реакцій між атомами парового потоку металу і атомами спеціально поданих у камеру хімічно активних газів. Такий процес називають реакційним. Найбільш поширеним прикладом такого процесу є формування покриттів із TiN при випаровуванні чи розпиленні в середовищі N2.
Для реакційного напилення покриттів необхідно знати, при якому тиску робочого газу буде забезпечено синтез потрібної сполуки. Для практичних завдань це можна вирішити таким чином.
Кількість атомів металу, що досягли поверхні напилення за одиницю часу, можна виразити через швидкість осадження (атом./(см2×с)) без врахування розсіяння газоподібними молекулами у вигляді:
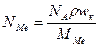 (13)
(13)
де N A – число Авогадро; r - густина матеріалу, г/см3; w k – швидкість конденсації металу, см/с; M Me – молекулярна (атомна) маса металу.
Кількість молекул газу, які стикаються з поверхнею напилення (моль/(см2×с)обчислюється за формулою:
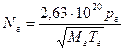 (14)
(14)
де р г – парціальний тиск газу, Па; М г – молекулярна маса газу; Т г – температура газу, звичайно Т г = 300 °С.
Наприклад, при отриманні TiN за реакцією Ті + N = TiN при швидкості конденсації wТік =6×10-6 м/с (М = 48, r = 4,5 г/см3) маємо NTi = 3,4×1017 ат/(см2×с)). Для отримання аналогічної частоти зіткнення молекул необхідно, щоб парціальний тиск становив p N2 = 0,1 Па. Таким чином, при тиску 10-1 Па буде утворюватись TiN.
Реакцію між атомами металу і газу можна стимулювати їх іонізацією. В іонізованому вигляді компоненти легше утворюють необхідні сполуки.
Вищі тиски газів у камері при реакційному напиленні порівняно з прямим випаровуванням або з розпиленням матеріалу підвищують імовірність зіткнення і дифузійного розсіяння конденсованих атомів та іонів.
Лекція 2.3.15. Поверхневі фізико-хімічні процеси. Структура і властивості конденсаційних покриттів.
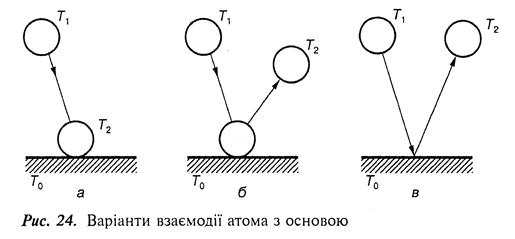
Випаруваний чи розпилений атом при зіткненні з основою потрапляє в поле дії сил атомів, розміщених на її поверхні. Після зіткнення можливі три варіанти поведінки атома (рис. 24):
1) адсорбується і залишається на поверхні;
2) адсорбується, але через деякий час знову відривається від поверхні, тобто відбувається повторне випаровування – ревипаровування;
3) ударяється об поверхню і відштовхується від неї.
Імовірність того, що атом утворює покриття, характеризує коефіцієнт конденсації – відношення кількості атомів, які конденсуються, до загальної кількості атомів, які досягають поверхні:
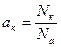 (15)
(15)
Після зіткнення з поверхнею атом обмінюється енергією. Обмін енергією характеризується коефіцієнтом термічної акомодації:
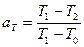 (16)
(16)
де Т 0 – температура поверхні основи; Т 1, Т 2 – середні еквіваленти температури відповідно падаючих і відбитих атомів.
Температури Т 1 і Т 2 розраховують як кінетичну енергію атомів:
 (17)
(17)
де w – середня швидкість; m – маса атома; k – стала Больцмана.
Якщо падаючий атом повністю віддає енергію основі (тобто Т 2 = Т 0), то а Т = 1, і між атомом і поверхнею встановлюється термічна рівновага. За відсутності обміну Т1 = Т2, і тоді а Т = 0, тобто завжди коефіцієнт термічної акомодації знаходиться в межах від 0 до 1.
На основі сучасних уявлень фізики твердого тіла розроблено модель з використанням класичного розв'язання рівняння Шредінгера для руху частинок у періодичному потенціальному полі з параболічною формою потенціальних ям (рис. 25).
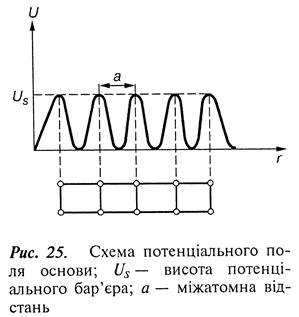
Якщо енергія атома велика, то процес її поглинання не проходить швидко і атом відбивається від поверхні, або, навпаки, якщо енергія мала, атом, не маючи її надлишку для переміщення по поверхні основи для знаходження стійкого положення, також відбивається від поверхні. Тому, виходячи з моделі, імовірність захоплення атома велика, коли його енергія не перевищує величину Еа» 25Ед (Ед – енергія десорбції).
Енергія десорбції приблизно становить Ед = 0,5Es, де Es – енергія сублімації.
Для моноатомної пари металів Ед= 1...4 еВ.
При зіткненні атом віддає свою енергію і прагне до теплової рівноваги з основою. Час, коли різниця температур адсорбованого атома і основи зменшується в е разів, називається часом релаксації і оцінюється співвідношенням t p»2/ v, де v – частота коливань адсорбованого атома.
Якщо термічна рівновага встановлюється, то час знаходження атома на поверхні відносно до часу релаксації визначається за формулою:
 (18)
(18)
При t p ³ t з відбувається десорбція (атом не прилипає).
Час знаходження атома на поверхні завжди набагато більший від часу релаксації навіть при високих значеннях температури основи (Т 0). Коли поверхня основи забруднена, то і в цьому випадку адсорбований атом не досягає термічної рівноваги і має температуру Т, вищу від температури поверхні основи (гарячий атом), а також велику імовірність ревипаровування.
Якщо є надлишок енергії, то атом на поверхні може переміщуватись.
Скупчення атомів, які затримуються в потенціальних ямах на відстанях, кратних міжатомним відстаням основи (рис. 26), є початком утворення центрів конденсації чи зародків нової фази.
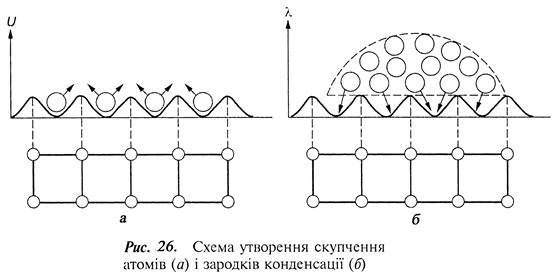
Між атомами в скупченні починають діяти сили хімічного зв'язку, які прагнуть стягнути ці атоми в положення, які визначаються їх щільним розміщенням у відповідній кристалічній ґратці (рис. 26, а).
Взаємодія адсорбованих атомів з атомами поверхні основи приводить до появи протилежно напрямлених сил, що прагнуть розтягнути скупчення (рис. 26, б). Поява скупчень атомів приводить до збільшення вільної енергії системи, а сили поверхневого натягу, що виникають на межі скупчення-пара і скупчення-основа, намагаються стягнути скупчення в компактне утворення – зародок, їм протидіють сили поверхневого натягу основи.
Утворення зародків при конденсації пари на поверхні основи можна розглядати з макроскопічних позицій (термодинамічний підхід) і з позицій міжатомної взаємодії адсорбованих атомів (статистичний підхід).
Якщо при конденсації виникає зародок з ізотропною енергією, то можна припустити, що він матиме куполоподібну форму (рис. 27).
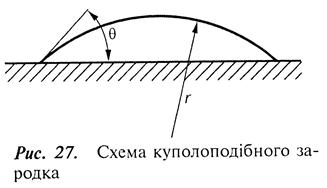
Термодинамічний та статистичний аналізи дають правильне значення швидкості утворення критичних зародків доти, доки не відбудеться насичення, тобто коли середня відстань між зародками не дорівнюватиме середньому дифузійному пробігу адсорбованого атома. З цього часу зародок розвивається шляхом захоплення дифундуючих атомів, що перешкоджає подальшому їх утворенню. Як наслідок критичний зародок переходить у закритичну стадію. Такий розрослий зародок прийнято називати острівцем.
Зародженням і ростом центрів конденсації починається формування покриття. Відзначають кілька стадій росту покриття при конденсації пари (рис. 28).
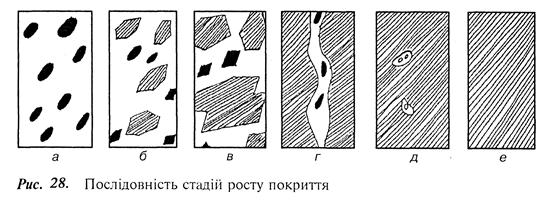
На першій стадії виникають хаотично розміщені тривимірні зародки (рис. 28, а), які через деякий час перетворюються на острівці (рис. 28, б), їх форма залежить від поверхневої енергії і умов конденсації. Розростання острівців зменшує відстань між ними, і в місцях стикання сусідніх острівців відбувається їх злиття – коалесценція (рис. 28, в). Внаслідок коалесценції формується розвинена мережа каналів, які не заповнені сконденсованою речовиною (рис. 28, г). Канали поступово зарощуються і перетворюються на окремі пори різноманітної форми (рис. 28, д). Кінцевою стадією є утворення суцільної плівки (рис. 28, е).
Масоперенесення при коалесценції відбувається внаслідок поверхневої дифузії, тому обмеженим фактором цієї стадії є поверхнева дифузія, а рушійною силою – мінімізація поверхневої енергії.
На зрощування острівців і формування покриття помітно впливає енергія атомів, що падають на поверхню основи. При відносно низьких температурах основи збільшення кінетичної енергії атомів пари, наприклад при переході від термічного випаровування до катодного, прискорює зрощення острівців. Це пояснюється суттєвим збільшенням дифузійної рухливості внаслідок додаткової кінетичної енергії адсорбованих атомів. Зрощення острівців прискорюється також із підвищенням температури основи, швидкостей випаровування і конденсації.
Формування конденсованої плівки супроводжується виникненням великої кількості лінійних недосконалостей – дислокацій. Їх щільність сягає 1010-1011 см-2. Більшість дислокацій виникає на остаточних стадіях утворення суцільної плівки. Характерним виявом великої кількості дислокацій у тонких плівках є розвиток субструктури, яка впливає на їх властивості.
Структура і властивості покриттів. Температура поверхні конденсації – один з основних параметрів, який визначає структуру покриття.
За результатами численних досліджень запропоновано тризонну модель структури покриттів (рис. 29).
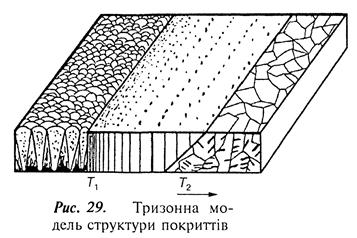
Перша зона – низькотемпературна, формується при температурі поверхні основи від кімнатної до деякої граничної температури Т 1» 0,3 Т пл конденсованої речовини. Поверхня покриття має куполоподібну будову нижчу від температури Т 1. У перерізі покриття спостерігаються конусоподібні кристали. У внутрішніх об'ємах кристалів і особливо в прилеглих зонах наявні мікропори.
Друга зона – проміжна між Т 1 і Т 2, де Т 2 = (0,45...0,50) Т пл конденсованої речовини. Поблизу Т 1 відбувається поступовий перехід до другої зони з рівною матовою поверхнею. У перерізі спостерігається стовпчаста структура. Особливістю структури є наявність міжкристалітних меж. Мікропористість покриттів у цій зоні практично не спостерігається. Ширина стовпчастих кристалів росте зі збільшенням температури основи.
Третя зона – високотемпературна, формується при температурах, вищих від Т 2. У цій зоні утворюється практично рівноважна структура. Якщо речовина здатна до поліморфного перетворення, то в покритті відбувається додаткова зміна структури при цій температурі.
Температурна залежність розмірів кристалітів D в кожній структурній зоні покриттів залежить від ефективної енергії активації процесів U, що контролюють формування кристалітів (зерен).
Низькі значення енергії активації, характерні для першої зони, відповідають значенням енергії активації переходу атомів між двома сусідніми положеннями рівноваги на поверхні Us. Реалізація тільки цих переходів сприяє виникненню куполоподібних зародків конденсації, які потім ростуть, утворюючи конусоподібні кристали.
У другій структурній зоні розвивається більш складний механізм росту кристалів, який складається з двох послідовних процесів: утворення критичних зародків і подальшого їх розростання по поверхні до стикання один з одним. Внаслідок цього утворюються досить досконалі покриття з упорядкованим розташуванням лінійних недосконалостей у вигляді границь чи субграниць стовпчастих кристалів.
У третій зоні внаслідок високої енергії активації розвиваються процеси збиральної рекристалізації та зростання зерна, що приводить до утворення рівноважної структури.
У товстих покриттях шари, які розміщені ближче до основи і тривалий час знаходяться при високих температурах, мають більш рівноважну структуру. Якщо нанесення покриттів здійснюється при температурі основи, близькій до T 2, то нижні шари матимуть рекристалізовану структуру, а верхні (холодніші) – стовпчасту.
У покриттях сплавів типу твердих розчинів спостерігається більш дрібнозерниста структура порівняно з покриттями із чистих металів.
У двофазних покриттях змінного складу завжди спостерігається подрібнення стовпчастих кристалів з одночасним збільшенням ступеня їх рівноважності. У загальному випадку залежність розміру зерна від об'ємного вмісту другої фази в покриттях має експоненціальний характер.
Конденсаційні покриття мають чітку залежність механічних властивостей від структури. Ця залежність визначається додатковим розмірним фактором. Особливо це спостерігається для двофазних матеріалів дисперсного типу, які поділяються на дві групи:
1) дисперсно-зміцнені матеріали; складаються з металевої чи керамічної матриці, в якій рівномірно розподілено 5-10 % некогерентних дисперсних частинок другої фази, як правило, з вищим модулем пружності порівняно з матрицею;
2) двофазні дисперсні матеріали; містять у собі 60-80 % частинок міцнішої другої фази.
Для першої групи матеріалів існує екстремум пластичності, коли середня вільна відстань між частинками дорівнює приблизно середньому розміру зерен матриці.
Другий екстремум механічних властивостей існує у двофазних конденсованих матеріалах, об'ємна частка яких становить 60-80 % другої фази. У таких структурах середня вільна відстань між частинками наближається до середньої відстані між частинками.
У композиціях, які містять у собі крихкі частинки другої фази у вказаному інтервалі об'ємної частки (60-80 %), одночасно з максимумом міцності спостерігається максимум пластичності чи в'язкості руйнування (для керамічних матеріалів).
Поверхні поділу між шарами (міжфазні поверхні) є джерелами далекодійних внутрішніх напружень і ефективними бар'єрами на шляху дислокацій, які рухаються. Тому мікрошаруваті матеріали можуть бути високоміцними при кімнатній температурі і жароміцними при високих температурах.
Товщина мікрошарів – один із найголовніших параметрів, які визначають властивості мікрошаруватих конденсатів. Різке підвищення мікротвердості спостерігається в конденсатах з товщиною мікрошарів меншою, ніж 2 мкм.
Останнім часом виділився ще один напрямок практичного використання так званих трансформованих мікрошаруватих матеріалів і покриттів для якісного дифузійного з'єднання окремих частин виробів, а також для виробництва виробів складної форми зі сплавів, які важко деформуються, наприклад інтерметалідів.
Сутність трансформованих мікрошаруватих матеріалів полягає в тому, що більшість сплавів на основі титану, нікелю, заліза та інших металів, які містять у собі компонент з низькою температурою плавлення, наприклад алюміній, можна за допомогою випаровування і конденсації отримати у вигляді мікрошаруватої композиції, що складається з мікрошарів легкоплавкого компонента, і сплаву без легкоплавкого компонента, які накладаються один на один.
При нагріванні мікрошаруватих матеріалів до температури вищої, ніж температура плавлення алюмінію, наприклад 700 чи 800 °С, проходить розплавлення мікрошарів алюмінію і розвиток фізико-хімічних реакцій, спочатку з участю рідкої фази, а потім і у твердій, що забезпечує трансформацію мікрошаруватих композицій в однорідний сплав заданого складу. Залежно від температури нагрівання і товщин мікрошарів час ізотермічної витримки, необхідний для отримання однорідного сплаву, можна варіювати в широких межах – від кількох хвилин до кількох десятків хвилин.
Нерівноважні процеси конденсації парової фази на основі створюють умови для розвитку різноманітних форм фізичної неоднорідності, зокрема мікропористості. Кількість, розміри, форма і просторовий розподіл пор залежить від хімічного складу парового потоку і умов конденсації (температури основи, швидкості конденсації, кута стикання атома чи молекули з основою та ін.).
На сьогодні існують експериментальні дані, які свідчать про можливість отримання товстих двофазних конденсатів із мікропористою, термічно стабільною структурою.
Особливості випаровування багатокомпонентних сумішей з одного джерела зумовлюють створення покриття з контрольованим вмістом кожного елемента за товщиною. Зміна складу парової фази залежно від тиску пари кожного елемента формує градієнт концентрацій за товщиною конденсату.
Якщо К < 1 (при К = 1 сплав випаровується узгоджено), то спочатку випаровується компонент із високим тиском пари. Потім зі збільшенням кількості сплаву, який випарувався, починається випаровування компонентів із низьким тиском пари. Ця відмінність тим сильніша, чим більше значення параметра К відрізняється від одиниці і чим вища вихідна концентрація компонента А. Внаслідок вказаних змін складу парової фази формується градієнт концентрацій за товщиною покриття. Прилеглі до поверхні конденсації шари містять у собі максимальну кількість компонентів з високим тиском пари.
Градієнтні матеріали із зовнішніми оксидними (А12О3, А12О3, MgO), карбідними чи боридними шарами перспективні як тверді і зносостійкі покриття.
Лекція 18. Закономірності газофазного осадження. Структура і властивості газофазних покриттів.
Закономірності осадження. При газофазному осадженні покриття утворюються внаслідок перебігу хімічних реакцій поблизу поверхні, на поверхні чи в приповерхневому шарі основи. Вихідними продуктами є газоподібні галогеніди, карбоніли чи металоорганічні сполуки, при розкладанні чи взаємодії яких з іншими газоподібними складовими сумішей (воднем, аміаком, вуглеводнями, оксидом вуглецю та ін.) можуть утворюватись і осаджуватись на оброблюваній поверхні потрібні матеріали.
Газофазне осадження здійснюють термічним розкладанням, відновленням, гідролізом, високотемпературним прямим окисненням.
При термічному розкладанні основу нагрівають до температури розкладання газоподібних робочих сполук з осадженням їхніх нелетких компонентів на поверхні основи. Процес здійснюється у вакуумі 10–1 Па, а також у нейтральному чи іншому підібраному середовищі газів-носіїв. Осадження простих речовин відбувається за реакціями
W(CO)6 ® W + 6СО,
ZrI4 ® Zr + 2І2,
В2Н6 ® 2В + ЗН2,
SiH4 ® Si + 2Н2,
оксидів –
Si(OC2H5)4 ® SiO2 + 4С2Н4 + 2Н2О,
2А1(ОС2Н5)3 ® А12О3 + 6С2Н4 + ЗН2О,
боридів –
2Ті(ВН4)3 ® 2ТіВ2 + В2Н6 + 9Н2.
Температури розкладання для різних робочих сполук коливаються у межах від 300 до 2300 °С і вище.
Леткі сполуки відновлюються воднем, аміаком та іншими речовинами, які містять у собі водень, а також парами металів.
Осадження простих речовин відбувається за реакціями
SiCl4 + 2Н2 ® Si + 4HC1,
WF6 + 3H2 ® 2W + 6HF,
2ВС13 + ЗН2 ® 2В + 6НС1,
МеГ+ 1/2Н2® Ме+ НГ.
Пари металів (К, Na, Mg, Zn, Cd) осаджуються аналогічно, наприклад
SiCl4 + 2Zn ® Si + 2ZnCl2.
Гідроліз газоподібних галогенідів водяною парою (Н2О) чи парою Н2 + СО2 використовується для отримання покриттів оксидів. У першому випадку водяна пара вводиться в систему безпосередньо, а в другому – утворюється внаслідок реакції
Н2 + СО2 ® Н2О + CO.
Осадження оксидів відбувається за реакціями
SіС14 + 2Н2О ® SiO2 + 4HC1,
2А1С1з + ЗН20 ® А12O3 + 6НС1,
SiCl4 + 2Н2 +2СО2 ® SiO2 + 4НС1 + 2СО.
Високотемпературне пряме окиснення киснем газоподібних галогенідів чи металоорганічних безкисневих сполук використовується для отримання покриттів з оксидів:
2А1С13 + 1,5О2 ® А12О3 + ЗС12,
ТіС14 + О2 ® ТіО2 + 2С12.
Аналогічно осаджується більшість інших елементів (Fe, Ni, Be, Al, Cr, Ті, Hf, Th, V, Nb, Mo, Та) та їхніх боридів, нітридів, карбідів і оксидів.
Газофазне нанесення покриттів умовно можна подати як послідовність елементарних процесів:
1. хімічне випаровування, пов'язане з отриманням металовмісних сполук;
2. перенесення газоподібної металовмісної речовини;
3. взаємодія газоподібної металовмісної речовини з поверхнею основи;
4. формування покриття.
Процес хімічного випаровування базується на зміні вільної енергії при утворенні леткої сполуки металу на відміну від термічного випаровування чи катодного розпилення, особливістю яких є передача кінетичної енергії, яка необхідна для випаровування чи розпилення. Тому хімічне випаровування може здійснюватися при нижчих температурах з економією енергії.
Із кінетичної теорії газів відомо, що підвищення температури металу приводить до зменшення концентрації адсорбованих атомів (наприклад, галогену), а підвищення тиску газової фази – до її зростання. При хімічному випаровуванні можливі два найважливіші випадки.
1. Швидкість реакції визначається адсорбцією галогену. Припустимо, що початкові швидкості w 2, w 3, w 4 великі порівняно з w 1. Тоді швидкість сумарної реакції буде пропорційною концентрації галогену, адсорбованого поверхнею. Оскільки швидкості інших процесів високі, галоген реагуватиме відразу після адсорбції. Отже, концентрація адсорбованого галогену ніколи не буде високою.
2. Швидкість реакції визначається виходом атомів металу з ґратки в положення, сприятливе для здійснення адсорбції. Припустимо, що початкові швидкості w 1, w 3, w 4 вищі, ніж w 2. Якщо галоген сильно адсорбується, високе значення w 1 приводить до того, що при тиску, вищому від мінімального, необхідного для насичення, майже всі доступні місця адсорбції будуть зайняті. Якщо атом чи молекула галогену віддаляється від поверхні внаслідок реакції з металом, вільне місце займає частинка з газової фази.
Отже, доти, доки тиск галогену вищий від певного мінімального значення, швидкість реакції не залежатиме від тиску.
Товщина дифузійного примежового шару може змінюватися не тільки за часом, а й за відстанню.
Хімічне осадження з газової фази за своєю природою є багатостадійним. При перевищенні критичного розміру зародок у процесі росту стає кристалом.
Структура і властивості газофазних покриттів. Структура газофазних покриттів може бути трьох типів, які є наслідком отримання покриттів при певному температурному режимі (низько-, середньо- і високотемпературному). Інтервал кожного з режимів залежить від природи осаджених матеріалів і в більшості випадків відповідає приблизно 0,1; 0,2; 0,3 температури їх плавлення (рис. 30).
Поверхня покриттів, отриманих у низькотемпературній ділянці, цілком складається з великих сфероїдів. Чим вища швидкість подачі пари металовмісної сполуки, тим при значно нижчій температурі утворюються великі сферичні скупчення металу на поверхні покриття.
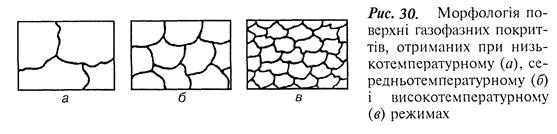
У середньотемпературній ділянці поверхня металевих покриттів має сфероїди другого типу з розмірами в 5–10 разів меншими, ніж розміри сфероїдів, які одержані в низькотемпературній ділянці.
У високотемпературній ділянці поверхня металевих покриттів складається з хаотично орієнтованих дуже малих кристалів, розміри яких у 10–15 разів менші, ніж розміри сфероїдів другого типу.
Таким чином, підбираючи технологічні параметри процесу, можна отримувати газофазні металеві покриття з потрібним рельєфом поверхні. Внутрішня будова покриттів, одержаних у низько-, середньо- і високотемпературних ділянках, має також три типи структури (рис. 31).
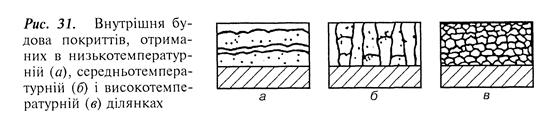
Внутрішня структура покриттів, отриманих у низькотемпературній ділянці, має горизонтально-шаровий характер будови. Покриття складаються лише з поперечних шарів з темними включеннями. Домішки концентруються переважно по межах зерен.
Другий тип структури утворюється за середнього інтервалу температур і різко відрізняється від першого. Покриття цього типу складаються з характерних вертикально-стовпчастих утворів, іноді – з горизонтальними примежовими шарами.
У високотемпературній ділянці утворюються покриття з дрібнокристалічною структурою. Цей тип внутрішньої структури нагадує третій тип мікрорельєфу поверхні покриттів, отриманих у цих же інтервалах температур.
Залежність швидкості наростання газофазних покриттів від температури основи характеризується кривою з максимумом (рис. 32).

Шаровий характер газофазних покриттів залежить від конкретних умов тепло- і масопередачі. У початковий період термічного розкладання виділення газоподібних продуктів спричинює зменшення концентрації металовмісної сполуки поблизу основи. Зростання швидкості виділення газоподібних продуктів розкладання і продуктів вторинних реакцій (вуглецю, карбідів, оксидів і нітридів) сприяє їх адсорбції на поверхні покриття, при цьому відбувається забруднення металу домішками. Видалення газоподібних продуктів і продуктів вторинних реакцій шляхом відкачування призводить до підвищення парціального тиску металовмісної сполуки в газовій фазі. Процес осадження інтенсифікується, і цикл повторюється.
Початкову ділянку визначають кінетичні фактори реакції термічного розкладання. Максимум швидкості зростання покриття досягається за умови рівності потоків – металовмісної речовини до основи і продуктів розпаду від неї. Вона сприяє утворенню стовпчастих кристалів.
Кінцеву ділянку визначає процес розкладання частини металовмісної сполуки, який збільшується при високих температурах реакційного газу.
Властивості газофазних покриттів оцінюються насамперед отриманням на основному металі поверхневого шару за умови найекономнішого легування. При збагаченні низьковуглецевої сталі хромом досягається корозіє- і окалиностійкість.
При газовому азотуванні, як і цементації, можна отримати тверді поверхневі зносостійкі шари з високою опірністю утомі. На маловуглецевих сталях можна отримати корозійностійкий шар хрому, а на матеріалах з більш високим вмістом вуглецю – зносостійкий шар карбідів хрому.
Лекція 19. Закономірності дифузійного утворення покриттів. Структура та властивості дифузійних покриттів.
Закономірності дифузійного насичення. Механізм дифузії є основою утворення дифузійних покриттів. Його схему можна розглянути на прикладі утворення покриттів із парогазової фази (рис. 33).

Рис.33 Схема утворення покриттів із паро газової фази
Із загальної кількості частинок N0 речовини А, які досягають поверхні тіла В за одиницю часу, одна частина Np ревипаровується (відбивається), друга Nк – конденсується і залишається в чистому вигляді на поверхні, третя Nд – поглинається тілом В. Поглинання відбувається внаслідок як дифузії А в В, так і зворотної дифузії – В в А. Швидкість зворотного процесу іноді може на два-три порядки перевищувати швидкість прямого процесу. Результуючу всіх процесів можна записати у вигляді рівняння:
N0 –Np = Nк + N д
Від швидкості кожного з цих потоків залежать товщина і характер покриття, яке утворюється (нашароване, дифузійне, комбіноване).
Якщо речовина А конденсується (осаджується) на холодній поверхні тіла В, і дифузія практично відсутня (N д = 0), то утворюється нашароване покриття з чітко вираженою межею поділу з основою (рис. 34, а).
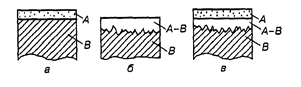
Рис. 34. Схема утворення нашарованого (а), дифузійного (б) і комбінованого (в) покриттів.
Якщо поверхня тіла В достатньо нагріта та активна і дифузійний потік дорівнює потоку речовини (N0 - Np = N д ), яка конденсується, або випереджує його, то утворюється типове дифузійне покриття (рис. 34, б). При цьому на поверхні тіла В створюється рівноважна концентрація речовини А, яка поглинається, що залежить від природи основи, природи і концентрації парогазового середовища, температури процесу, зовнішнього тиску та інших факторів.
Якщо поверхня тіла В недостатньо нагріта і дифузійний потік відстає від потоку речовини, яка конденсується (N0 - Np > N д ), то утворюється покриття комбінованого типу (рис. 34, в), де верхній шар складається з чистої речовини А, а нижній – із різного поєднання речовин А і В.
Таким чином, для отримання дифузійних покриттів необхідно, щоб основа добре розчиняла матеріал покриття, який наноситься, і температура основи була достатньою для інтенсивного проходження дифузійних процесів.
Основними стадіями формування покриттів є:
1) очищення поверхні від оксидних плівок (їх розчинення, відновлення);
2) отримання елемента покриття в атомарному стані (з пари, газової, рідкої чи твердої фаз);
3) перенесення елемента покриття в будь-якому транспортувальному середовищі (газ, пара, рідкий розчин);
4) адсорбція на поверхню й утворення зародків кристалічної структури зовнішнього шару покриття (твердий розчин або сполука);
5) зміна складу і структури в прилеглому об'ємі основного металу, а також ріст шарів покриття (формування одно- або багатокомпонентних покриттів у перехідній зоні).
Очищення поверхонь можливе шляхом відновлювальних реакцій при взаємодії з активаторами, які вводяться в насичувальне середовище (наприклад, галогеніди, водень), чи безпосередньо із середовищем (розчини солей); розчинення плівки кисню в металі (дисоціація плівки внаслідок дифузії кисню в основу); сублімації в насичувальне середовище.
При насиченні з твердої фази виріб безпосередньо контактує з дифундуючим елементом (так званим дифузантом), який знаходиться в твердій фазі. У цьому випадку використовують подрібнені чисті метали чи феросплави у вигляді порошку. Надходження дифузанту в поверхневі шари здійснюється лише через площину фактичного контакту металів у твердій фазі. При цьому необхідне встановлення загального металевого зв'язку між металом виробу і металом покриття. Дискретність такого контакту приводить до поверхневої дифузії, яка надалі переходить в об'ємну дифузію внаслідок поповнення елементів із точок безпосереднього контакту. За цією схемою отримують дифузійні покриття на залізі і хромонікелевих сталях та сплавах при насиченні вольфрамом, молібденом, ніобієм, танталом. Такий метод застосовується у випадках, коли пружність парів елементів значно нижча за пружність пари основи.
При насиченні з рідкої фази дифузант може потрапляти з розплавів легкоплавких металів (Al, Cu, Zn, Cd, Pb, Sn та ін.) або з розчинів солей. Прикладом першого варіанта є гаряче цинкування й алітування в розплавах цих металів. При гарячому цинкуванні в розплавленому металевому цинку при температурі 430-460 °С протягом кількох хвилин забезпечується товщина дифузійного шару 0,02-0,03 мм.
Насиченням із розчинів солей отримують покриття хрому, бору, алюмінію. Необхідний елемент вводиться в склад сольової ванни у вигляді металу або солі металу, який насичується. Процес відбувається при ізотермічному витримуванні у ванні з електролізом або без нього.
У загальному випадку при насиченні з розчинів солей в атомарний стан дифузанту переходить внаслідок хімічних обмінних реакцій на зовнішній міжфазній межі середовище – виріб. При насиченні з газової фази атомарний стан дифузанту може бути результатом реакцій відновлення чи термічного розкладання і його особливості подібні до газофазного осадження. Стадія перенесення в транспортній фазі не обмежує процес і здійснюється внаслідок конвекції.
На межі шар – метал за умов досягнення квазірівноважного стану можливе виникнення двох основних ситуацій:
1) насичене середовище внаслідок перебігу процесів і реакцій відновлення генерує елемент покриття (дифузант) в атомарному стані;
2) якщо утворення дифузанту в атомарному стані відбувається внаслідок обмінних реакцій компонентів газової фази, які містять у собі дифузант з насиченим металом, то рівноважна концентрація дифузанту в адсорбованому шарі Ср не може бути більшою за Cs, оскільки після досягнення в поверхневому шарі концентрації Cs встановлюється стаціонарний процес, який підтримує рівність хімічних потенціалів дифузанту між фазами.
Процес утворення сполуки з твердого розчину можна поділити на два періоди: інкубаційний і кінетичний. В інкубаційному періоді в окремих місцях поверхні накопичуються більш рухливі елементи покриття й основи, що приводить до формування центрів виділення сполук. Якщо елементи насичення і конструкційний метал утворюють кілька сполук, то в окремих ділянках виділяються сполуки різного складу. При певній їх концентрації ґратки твердих розчинів перебудовуються в ґратки цих сполук. При цьому вже в кінетичний період межа сполука – твердий розчин пересувається шляхом утворення на окремих ділянках пересиченого твердого розчину, тобто вона пересувається усереднено. Після утворення щільного шару сполуки відбувається масоперенесення до основного металу крізь шар сполуки.
У випадку обмеженої розчинності системи елемент покриття – основа досягнення концентрації насичення призводить до перебудови кристалічної ґратки твердого розчину.
У практиці дифузійного насичення розраховують ефективний коефіцієнт дифузії, як правило, для багатокомпонентного сплаву.
Коефіцієнт дифузії залежить від багатьох факторів: температури, концентрації, кристалічної структури, розмірів атомів дифузанту та ін. Наприклад, можна прийняти, що температурна залежність коефіцієнта дифузії описується законом Арреніуса для термічно активованих процесів:
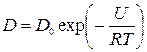 (1)
(1)
де D0 – передекспотенціальний множник, U – енергія активації дифузії.
Для самодифузії енергія активації U» 34 Tпл, де Tпл – температура плавлення дифузанту.
Час, необхідний для отримання покриття заданої товщини при сталому коефіцієнті дифузії, можна визначити так:
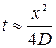 (2)
(2)
де х – координата в напрямку потоку.
Звідси випливає, що (2) може бути використано для оцінювального розрахунку часу насичення на певну глибину.
Якщо при насиченні утворюється кілька шарів, то на межі між ними відбувається стрибок концентрації елемента насичення. Таку дифузію називають реактивною (реакційною). Для її оцінки необхідно знати коефіцієнти дифузій у фазах і швидкості їх зростання.
Структура і властивості. Під час дифузійного насичення при отриманні покриттів важливо знати характер утворення і росту шарів, а також кінетичні параметри, які характеризують зміну концентрації елемента насичення за глибиною покриття в часі.
Можливість створення дифузійних покриттів визначається насамперед різницею діаметрів атомів металу основи. Наприклад, при дифузії в залізо елементів з великим діаметром атомів така різниця не повинна перевищувати 15-16 %, інакше напруження, які виникають у кристалічній ґратці заліза, будуть перевищувати границю її пружної стійкості. Ґратка стає нестійкою і виключає можливість дифузійного проникнення таких великих атомів у гратку заліза. Крім цього, елемент, який наноситься, має розчинятися в металі основи при кімнатній і підвищеній температурах.
Для підвищення зносостійкості використовують різні способи насичення, які поділяють на дві групи: насичення хімічними елементами (однокомпонентні, двокомпонентні і багатокомпонентні покриття); насичення хімічними сполуками (карбідами, нітридами, оксидами) (рис. 35).

Рис. 35. Схема видів покриттів чорних металів і сплавів насиченням хімічними елементами
Розглянемо поширене насичення поверхонь бором. Бор наежить до елементів з невеликим атомним радіусом (0,091 нм). Він вільно дифундує в залізо (атомний радіус a-Fe 0,24 нм) і подібно до вуглецю та азоту може створювати твердий розчин вкорінення. Розчинність бору в a-Fe і g-Fe невисока і становить відповідно від 0,004 % при температурі 710 °С до 0,008 % при температурі 906 °С і від 0,008 % при температурі 906 °С до 0,021 % при температурі 1149 °С.
Зміцнення при боридуванні металів і сплавів відбувається внаслідок утворення на оброблюваній поверхні металоподібних сполук – боридів. Ці сполуки називають металоподібними, тому що, крім нехарактерних для металів властивостей (дуже висока твердість і незначна здатність до пластичних деформацій), бориди мають властивості, характерні для металевого стану речовини, – високі електро- і теплопровідність, термоемісію, металевий блиск. Насичення бором значно підвищує поверхневу твердість, жаростійкість і корозійну стійкість виробу.
Лекція 20. Механізм осадження і закономірності формування гальванічних покриттів.
Гальванічні покриття отримують електроосадженням на поверхні основи, яка є провідником. Метал, на який наноситься покриття, занурюється в електропровідний розчин, що містить у собі солі цього металу. Катодом є основний метал (виріб), а анодом – стрижень або лист металу, який буде покриттям. У цьому випадку матеріал анода переходить у розчин, як тільки на катоді здійснюється його осадження, і, таким чином, підтримується концентрація іонів металу в розчині. Можна використовувати анод інертного матеріалу. У цьому разі концентрація іонів металу в розчині підтримується добавлянням відповідних солей у міру перебігу процесу електролізу (рис. 36).
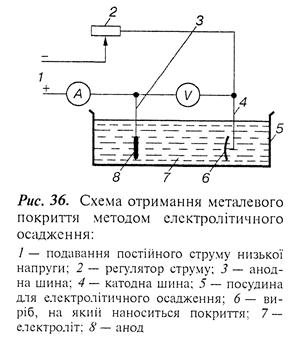
Електроосадження є катодним процесом звичайної електрохімічної реакції, яка викликає корозію на аноді. Реакція проходить під контролем складу електроліту, а потенціал і густина струму повинні мати такі значення, при яких здійснюється катодне відновлення іонів металу. У зв'язку з цим метал більшою мірою осаджується, ніж анодно окиснюється до утворення катіонів або інших окиснених форм.
Гальванічні покриття можуть бути з чистих металів, сумішей металів, сплавів або металів, які змішані з неметалевими речовинами, їх називають композиційними електрохімічними покриттями (КЕП).
Осад чистого металу отримується з електролітів, до складу яких входять солі цього металу. Для утворення осадів із металів і сплавів потрібні електроліти, які містять у собі солі складових цих матеріалів, які осаджуються або окремо, або безпосередньо у вигляді сплаву.
Неметалеві осади виникають при використанні розчинів, в яких неметали містяться у вигляді простих суспензій або сполук, здатних до руйнування і осадження на катоді.
У водних розчинах відновлення іонів водню до газоподібного стану можливе при відновленні катіонів або складних аніонів, які осаджуються на металі. Чим від'ємнішим є потенціал системи Me2+–Me, тим більша тенденція до виділення водню. Цинк та марганець, які мають порівняно з іншими металами найвід'ємніші потенціали, використовуються для отримання електролітичних покриттів. Для металів із ще від'ємнішим потенціалом слід використовувати розплавлені солі або розчини, які не містять у собі води.
Механізм осадження і закономірності формування покриттів. Осадження покриття починається з утворення центрів кристалізації в місцях порушення кристалічної ґратки основного металу (наприклад, структурних дефектів на поверхні) з наступним ростом кристалів металу, що осаджується, у місцях їх утворення. Так само на поверхні виробу досягають зростання зв'язного кристалічного металевого покриття, яке зчеплюється з металом атомними зв'язками, що забезпечує адгезію без утворення шарів проміжних сплавів між покриттям і основою (якщо між металами, що використовуються в твердому стані, неможлива дифузія при температурі, необхідній для електроосадження, або при наступних зберіганні чи експлуатації).
На початковій стадії утворення осаду в кристалічній структурі покриття можуть бути дефекти (наприклад, пори). За умови, що робочі параметри процесу електроосадження забезпечують оптимальні умови для зростання осаду, ці дефекти усуваються при збільшенні товщини осаду. Як правило, пори зникають, якщо товщина покриття досягає кількох мікрометрів.
Електроосадження металів відбувається за законами Фарадея: маса металу, що утворюється при електролізі, прямо пропорційна кількості електрики, яка пропускається крізь розчин, і хімічному еквіваленту металу. Таким чином, середню товщину покриття, що осаджується, можна легко розрахувати, знаючи силу струму, час нанесення покриття, площу поверхні, яка обробляється, і хімічний еквівалент металу, який використовується для покриття. Існує ще один додатковий фактор, який впливає на результати розрахунку: ККД катода під час електроосадження. Наприклад, для міді значення ККД приблизно дорівнює 100 %, якщо осадження здійснюється з кислотного розчину сульфату міді. Однак для іншого розчину чи металу ККД може значно зменшуватись і досягати, наприклад, для хрому 8–18 %. Крім того, товщина осаду залежить від відстані між анодом і катодом. Здатність розчину електроліту при нанесенні гальванічних покриттів долати цю залежність називається його розсіювальною здатністю (або макророзсіювальною здатністю). Наприклад, мідь – метал із доброю розсіювальною здатністю, хром – з поганою. На цю властивість може впливати також склад ванни і режим її роботи. Через обмежену розсіювальну здатність на товщину осаду впливає форма поверхні виробу, який обробляється: товщина осаду збільшується на гострих крайках та виступах і незначно зменшується в заглибленнях і впадинах.
Здатність електроліту знижувати ступінь шорсткості на поверхні основного металу, тобто його мікророзсіювальна здатність, є особливою властивістю, що називається вирівнюванням. Електроліт із позитивними властивостями вирівнювання створює осад, який поступово вирівнюється на поверхні основного металу в міру збільшення товщини шару покриття. Вважається, що різниця в поляризації мікропіків та мікрозаглиблень на поверхні основного металу впливає на співвідношення швидкостей дифузії іонів і адсорбції на поверхні, локально змінюючи швидкість електроосадження.
Властивості вирівнювання зазвичай контролюються введенням спеціальних домішок в електролітичну ванну. Домішки є органічними сполуками (наприклад, кумарин у розчині для нанесення нікелевого покриття). Здатність до мікровирівнювання і розсіювання часто поєднуються в одному розчині, але це не обов'язково, наприклад, у цинку добра розсіювальна здатність, але погана здатність до вирівнювання.
Якщо електроосадження здійснюють із використанням простих розчинів солей металу, то зазвичай отримують матові осади. У таких випадках досягти блискучих покриттів можна лише внаслідок полірування і глянсування, однак ці операції є тривалими і дорогими.
Крім того, блискучі покриття можна отримати безпосередньо після обробки у ванні, вводячи особливі присадки до складу електроліту. Для цього зазвичай використовують поверхнево-активні речовини і колоїди, які сприяють комплексному утворенню іонів металу та впливають на адсорбцію і локалізовану катодну поляризацію. Також вони можуть впливати на процес кристалізації осадів (про це свідчить, наприклад, шарувата мікроструктура блискучого покриття нікелю порівняно зі стовпчастою мікроструктурою матового нікелевого покриття). Блискучі покриття отримують тільки при обмеженій густині струму (яка змінюється також під дією особливих присадок), тому матова поверхня утворюється на крайках фігурних виробів, де під час нанесення покриття досягається найбільша густина струму.
Опірність корозії зменшується зі збільшенням внутрішнього механічного напруження внаслідок підвищення здатності гальванічного покриття до руйнування в міру розвитку корозії. При порушенні захисних властивостей покриття основний шар залишається незахищеним. Внутрішні напруження в покритті можуть бути зумовлені структурною невідповідністю між основним металом і найближчими до нього атомними шарами покриття або між способом осадження і кристалізацією металу з електроліту.
На структурну невідповідність не можуть впливати умови нанесення покриття. Внутрішнє напруження можна зменшити лише додатковим введенням іншого покриття між основним матеріалом і вибраним металом, щоб структурна різниця розподілялась між двома міжфазними межами. Напруження внаслідок електроосадження і кристалізації дуже часто змінюється залежно від складу електроліту або параметрів осадження. Наприклад, матові нікелеві покриття мають низьке внутрішнє напруження, а блискучі – високе.
Особливістю гальванічного процесу є виділення водню на катоді. Молекули водню, які отримані відновленням іонів водню або молекул води, можуть виділятися в газоподібному стані, а водень в атомарному стані може дифундувати або в покриття, або в основний метал. Виділення на катоді водню у вигляді газових бульбашок може стати причиною утворення порожнин, нерівних осадів або осадів із кристалічними дефектами. З метою усунення цього газ з поверхні катода видаляється перемішуванням розчину або переміщенням катода.
Якщо водень, який виділяється на катоді, поглинається покриттям або основним металом, то це може зумовити крихке руйнування (наприклад, руйнування високоміцних сталей під час покриття цинком або кадмієм). У таких випадках термічна обробка після нанесення покриття дасть змогу видалити водень і запобігти утворенню тріщин.
Перспективним напрямом розвитку технологій гальванічних покриттів є композиційні електрохімічні покриття (КЕП).
Композиційні електрохімічні покриття складаються з металевої матриці і тонкодисперсних частинок іншої фази, розподілених в її об'ємі. Розмір таких частинок становить 0,01–50 мкм, об'ємна частка – 1–50 %.
Як дисперсну фазу використовують порошки окремих елементів, але найчастіше застосовують хімічні сполуки на органічній чи неорганічній основі – безкисневі тугоплавкі сполуки, оксиди, полімери та ін.
Тонкодисперсні частинки в необхідній кількості вводять в електрохімічні ванни, які в процесі електролізу осаджуються на катоді і зарощуються іонами металів, що виділяються на ньому.
Залежно від властивостей і кількості дисперсної фази КЕП можуть мати високі твердість і зносостійкість, жароміцність, жаро- і корозійну стійкість, анти- і фрикційні властивості та інші необхідні експлуатаційні характеристики.
Композиційні комбіновані електролітичні покриття поділяються на одношарові (рис. 37, а) і багатошарові (рис. 37, б, в).
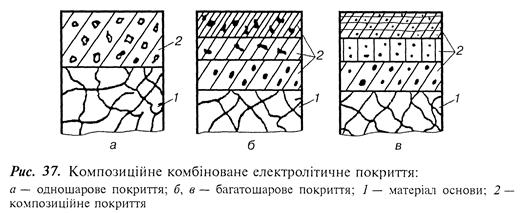
За характерними ознаками і призначенням КЕП поділяють на моно- і поліметалеві, моно- і полікомпозиційні, тонко- і товстошарові, моно- і поліфункціональні.
КЕП отримують із суспензій у вигляді електропровідних рідких розчинів із додаванням високодисперсних твердих частинок чи з емульсій, які утворюються при введенні в електроліти гідрофобних рідин або середовищ, що утворюють піни.
При проходженні крізь електроліт електричного струму на поверхні деталі (катоді) осаджуються метал чи сплав (матриця покриття) і тонкодисперсні частинки (друга фаза), які зарощуються і цементуються матрицею.
Осадження КЕП проводять при безперервному перемішуванні суспензії з метою забезпечення розміщення частинок у завислому стані і рівномірного їх осадження на поверхню.
Механізм утворення КЕП визначається двома основними факторами: складом і властивостями електроліту-суспензії (EC) та умовами електролізу. Ці фактори складаються, як правило, з незалежних параметрів, а іноді – параметрів, які взаємно впливають один на одного. До таких параметрів належать: для EC – склад електроліту, значення рН середовища, температура, концентрація дисперсних частинок в об'ємі EC, їх природа, розмір і форма; для умов електролізу – вид електричного струму (постійний, нестаціонарний – реверсивний, імпульсний, асиметричний), його густина, спосіб перемішування EC та ін. Цими параметрами визначаються термодинаміка і кінетика процесу осадження КЕП.
За певних умов електролізу можливе отримання покриттів, що містять у собі частинки від мінімального розміру до частинок, розмір яких близький до товщини
|
|
|
|
|
Дата добавления: 2014-01-11; Просмотров: 3704; Нарушение авторских прав?; Мы поможем в написании вашей работы!