КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Коагуляция коллоидных примесей воды
|
|
|
|
Если очистка воды от тяжелых грубодисперсных примесей может быть осуществлена обычным отстаиванием, то выделение коллоидно-дисперсных веществ из воды требует применения процесса коагуляции. Под коагуляцией понимают физико-химический процесс слипания коллоидных частиц и образования грубодисперсной макрофазы (флокул) с последующим ее выделением из воды.
Коллоидные частицы имеют весьма малые размеры и поэтому участвуют в броуновском движении, в то же время они обладают заметной скоростью диффузии (10-1 — 10-3 см2/с), что способствует выравниванию концентрации частиц по объему. Коллоидные системы обладают избытком свободной энергии за счет чрезвычайно развитой удельной поверхности частиц. Термодинамически такая система должна самопроизвольно стремиться к состоянию, в котором ее свободная энергия была бы минимальна, т. е. к самопроизвольному уменьшению поверхности, а следовательно, и к укрупнению частиц. Однако на практике коллоидные системы обладают весьма высокой агрегативной устойчивостью. Такая устойчивость при малых размерах частиц способствует седиментационной устойчивости (постоянству концентрации примесей по всему объему воды), так как гравитационная сила, вызывающая седиментацию, нивелируется силами диффузии. Агрегативная устойчивость коллоидной системы объясняется существованием двойного электрического слоя ионов и скачка потенциала на границе раздела фаз.
Двойной электрический слой возникает на поверхности частиц вследствие разных диэлектрических свойств дисперсной фазы и среды, воздействия молекулярных сил, обеспечивающих избирательную сорбцию ионов, или частичной диссоциации поверхностных молекул вещества частиц. Ионы, располагающиеся на поверхности частицы, называются потенци-алообразующими. Вследствие появления заряда на частице вокруг нее концентрируются ионы противоположного знака заряда — противоионы. Концентрация противоионов максимальна у поверхности (плотный ионный слой) и убывает с увеличением расстояния от поверхности частицы (внешний диффузный слой). Это объясняется одновременным действием как сил притяжения (электрических и молекулярных), так и диффузионных сил, стремящихся к выравниванию концентрации ионов в объеме воды.
|
|
|
Теоретически диффузный слой ионов распространяется на весь объем воды, практически же его толщину определяют дебаевской длиной X. Так, для симметрично-валентного электролита в предположении, что один вид ионов сорбируется на поверхности частицы, а другой составляет противоионы [1],
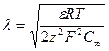 (1)
(1)
где F —постоянная Фарадея, равная 96 487 Кл/моль; R — универсальная газовая постоянная, равная 8,3143 Дж/(моль • К); z—заряд ионов; ε—диэлектрическая проницаемость среды, Ф/м; С∞,—концентрация ионов в глубине раствора, моль/л.
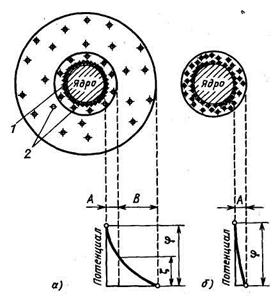
Рисунок 2.1 - Строение мицеллы:
а —ζ-потенциал больше нуля; б —изоэлектрическое состояние: ζ -потенциал равен нулю; 1 —потенциалообразующие ионы; 2 —противоионы; А — адсорбционный слой; В —диффузный слой
Значение величины X непостоянно и, как видно из формулы (1), зависит от концентрации раствора, заряда ионов, температуры. Например, в случае нейтральной воды при 298 К (Сн+= СОН-= 10-7 г-ион/л) дебаевская длина λ = 9,6 нм, а в 0,1 молярном растворе одно-одновалентного электролита X = 0,96 нм.
Таким образом, вокруг частицы возникает двойной электрический слой, включающий потенциалообразующие ионы и противоионы (рис. 2.1). В спокойном состоянии комплекс частицы с двойным электрическим слоем (мицелла) электронейтрален.
При движении частица увлекает с собой только часть прилегающего к ее поверхности слоя воды. Внешняя часть диффузного слоя остается вне сферы действия частицы. Это приводит к появлению электрокинетического потенциала, или ζ -потенциала, между частицей и раствором. Значение ζ -потенциала зависит от количества противоионов, увлекаемых с частицей; с их увеличением ζ -потенциал уменьшается. Повышение концентрации противоионов в диффузном слое должно приводить к увеличению их концентрации в плотном слое и, следовательно, к снижению ζ -потенциала. Более того, повышение концентрации противоионов в диффузном слое может привести к перезарядке частицы (изменяется знак заряда). Естественно, что при этом существует определенная концентрация противоионов, при которой ζ -потенциал становится равным нулю.
|
|
|
Коллоидные частицы, находящиеся в природных водах (песок, глинистые вещества, гуминовые кислоты), в основном приобретают заряд за счет диссоциации поверхностных молекул. Так как, эти вещества амфотерны, то вид и степень их диссоциации зависят от значения рН раствора. Значение рН, при котором эти вещества не диссоциируют, называется изоэлектрическим. При изоэлектрическом значении рН значение ζ -потенциала равно нулю. Для примесей природных вод значение изоэлектрических рН находится в кислой области. Так, для глины рНи э = 5, для гуминовых веществ рНиэ = 3,5-н4,5. Так как рН природной воды обычно равен 6,5 ч-8,5, то коллоидные примеси диссоциируют как кислоты с приобретением отрицательного знака ζ -потенциала частиц относительно раствора. Таким образом, в природной воде основная масса коллоидных частиц имеет одинаковый отрицательный заряд. Кроме того, частицы глины и гумуса способны к адсорбции ионов, причем последняя понижает их устойчивость к агрегации. В наибольшей степени понижают устойчивость трехвалентные ионы Fe3+ и А13 +. Ионы, образующие двойной электрический слой, способствуют удержанию молекул воды около частиц и возникновению в связи с этим гидратного слоя, препятствующего столкновению частиц друг с другом.
Агрегативная устойчивость дисперсных систем зависит от характера сил, действующих между частицами. На частицы, имеющие одинаковый знак заряда, действуют одновременно молекулярная сила притяжения (Ван-дер-Ваальсова сила) и электростатическая сила отталкивания.
|
|
|
Молекулярная сила притяжения двух сферических частиц радиусом г, находящихся на расстоянии R (между их центрами) друг от друга, описывается так:
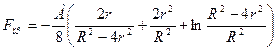
где А — константа Гамакера, приблизительно равная 10-6 Вт. При сближении частиц до очень малого расстояния эта зависимость упрощается:
Fnp=-Ar/(12H0), (2.3)
где H0 = R — 2r — расстояние между крайними точками сближающихся частиц.
При условии, что H0<.r, сила отталкивания таких частиц определяется уравнением Дерягина—Ландау
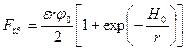
На рисунке 2.2 показан характер изменения сил притяжения и отталкивания в зависимости от расстояния между частицами. Так как законы изменения сил отталкивания и притяжения различны, результирующая сила имеет два энергетических минимума (потенциальные ямы), при достижении которых возможно сцепление частиц друг с другом. Правый минимум обеспечивает «дальнюю» коагуляцию с менее прочной связью между частицами. Более прочная связь наблюдается в левой потенциальной яме («ближняя» коагуляция), однако для этого сближающиеся частицы должны обладать достаточной энергией для преодоления энергетического барьера (максимум на результирующей кривой). Дальнейшему сближению частиц препятствуют гидратированные слои воды на их поверхности.
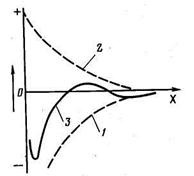
Рисунок 2.2 - Потенциальные кривые взаимодействия между частицами: 1 —энергия отталкивания; 2 —энергия притяжения; 3 — результирующая кривая
В природной воде коллоидные частицы не обладают достаточной энергией для преодоления энергетического барьера, а энергия сцепления в правой потенциальной яме недостаточна для их удержания в комплексе; этим объясняется их повышенная агрегативная устойчивость. Для повышения возможности сцепления частиц необходимо снизить силу отталкивания путем уменьшения значения ζ-потенциала. Экспериментально установлено, что при снижении ζ -потенциала до 0,03 В начинается процесс сцепления частиц, т. е. процесс коагуляции. Для этого достаточно ввести в воду сильный электролит, диссоциация которого позволит увеличить количество противоионов в двойном электрическом слое с соответствующим снижением значения ζ -потенциала.
|
|
|
Однако такой процесс возможен лишь при очень низких рН, что вызывает значительные неудобства на практике, связанные с защитой оборудования от коррозии, и, кроме того, приводит к повышению солесодержания воды. Поэтому при подготовке добавочной воды применяется процесс, основанный на взаимной коагуляции коллоидов, для чего в воду вводятся реагенты, образующие в ней коллоидный раствор с положительно заряженными частицами. Это нарушает устойчивость коллоидной системы и приводит к укрупнению частиц, образующих ее.
В качестве реагентов, называемых коагулянтами, обычно применяют сернокислые соли A12(SO4)3 и FeSO4- Эти соли в воде почти полностью диссоциируют:
Al2(SO4)3 →←2Al3 + + 3SO42-; (2.5)
FeSO4→←Fe2++SO42-. (2.6)
Катионы слабых оснований Аl3+ и Fe2+ легко подвергаются гидролизу. Так, гидролиз ионов А13+ протекает следующим образом:
А13++Н2О→←А1(ОН)2++Н +; (2.7)
А1(ОН)2+ + Н2О→←А1(ОН)2++Н +; (2.8)
А1(ОН)2++Н2О→←А1(ОН)3 + Н+ (2.9)
а гидролиз ионов Fe2 +:
Fe2 + +H2O→←Fe(OH)++H +; (2.10)
Fe(OH)++H2O→←Fe(OH)2 + H +. (2.11)
В щелочной среде (рН>8) и при достаточном количестве кислорода гидрат закиси железа окисляется в менее растворимый гидрат его окиси:
4Fe(OH)2 + O2 + 2H2O→←4Fe(OH)3. (2.12)
Как видно из приведенных реакций, достаточно полный гидролиз ионов А13+ и Fe2+ возможен лишь при условии отвода ионов Н +. В природной воде связывание этих ионов происходит согласно реакции
Н++НСОз- →←Н2СО→←СО2 + Н2О, (2.13)
поэтому наличие бикарбонат-ионов в воде является необходимым условием для обеспечения глубокого протекания процесса гидролиза. При недостаточной величине щелочности концентрация ионов Н+ может регулироваться введением NaOH, но это приводит к увеличению солесодержания воды. Таким образом, как видно из реакции гидролиза, его глубина существенно зависит от рН воды. При рН>7,5 образуется только А1(ОН)3, при более низких значениях рН получаются также А1(ОН)2 + и А1(ОН), которые затем, соединяясь с сульфат-ионами, образуют труднорастворимые соединения средних солей алюминия: A12(OH)4SO4—при рН ближе к 7 и A1(OH)SO4 — при рН ближе к 5,5. Эти соединения в интервале значений рН = 5,5-7,5 образуют коллоидный раствор с весьма малым значением ζ-потенциала. При рН<5,5 гидроокись алюминия растворяется полностью, а при рН>8 образуются ионы А1О2-.
Таким образом, при коагуляции ионы при помощи A12(SO4)3
необходимо поддерживать рН в интервале 5,5-7,5. В воде,
не содержащей посторонних ионов, изоэлектрическое значение (рН для А1(ОН)3 составляет 7,6-8,2, и при их наличии уменьшается на 1 —1,5 вследствие адсорбции ионов на поверхности частиц. Образование Fe(OH)3 происходит достаточно полно и быстро лишь при рН>8, для чего необходимо дозировать в воду совместно с коагулянтом щелочь или сочетать коагуляцию с процессом известкования.
Процесс коагуляции имеет две стадии: скрытую и явную. На скрытой стадии происходит формирование коллоидного раствора гидроксидов А13+ или Fe3+ и образование микрохлопьев. Именно на этой стадии коагуляции вода в основном и очищается от первичных коллоидных примесей. А затем на второй стадии процесса образуются крупные хлопья (флокулы) размером 1—3 мм, которые, обладая высокой сорбционной способностью, могут дополнительно извлекать примеси из воды.
При очистке природных вод в процессе коагуляции одновременно участвуют примеси различной степени дисперсности (в том числе и грубодисперсные) и различной природы (органические и неорганические). Такой процесс называется гетероадакоагуляцией. Отличительной его особенностью является преимущественная зависимость процесса от значения заряда частиц, заряженных слабее. Такие частицы могут слипаться не только между собой, но и с частицами, имеющими более высокий ζ-потенциал, вовлекая последние в процесс коагуляции. Роль слабозаряженных частиц и выполняют гид-роксиды алюминия и железа.
Образование макрофазы при коагуляции отличается от образования таковой при кристаллизации солей из пересыщенных растворов. Коагулирующий гидроксид сорбируется на поверхности грубодисперсных частиц и одновременно образует «клеевые мостики», связывающие эти частицы между собой в комплексы (рисунок). Органические вещества (например, гуминовые кислоты) сорбируются на поверхности коагулирующей гидроокиси. Образующаяся макрофаза (флокулы или хлопья) имеет весьма рыхлую структуру, так как промежутки между частицами заполнены водой. Она имеет плотность (1,001— 1,1 т/м3), мало отличающуюся от плотности воды, и невысокую механическую прочность. Размер вполне сформированных флокул составляет 1—3 мм. Эти флокулы затем выделяются из воды в процессе осветления.
В настоящее время теоретические разработки не дают оснований для вычисления необходимой дозы коагулянта для эффективного проведения процесса, поэтому доза подбирается экспериментально. Экспериментально подбирается и оптимально необходимое значение рН. Доза коагулянта зависит от состава примесей воды. Так, при большой концентрации грубодисиерсных веществ и малой концентрации коллоидных примесей доза коагулянта должна быть максимальна, и наоборот.
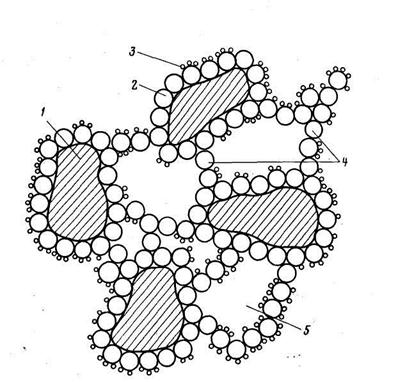
Рисунок - Структура хлопьев, образующихся при коагуляции:
1 — частицы ГДП; 2 —частицы гидроокиси; 3 —органические вещества; 4 —«клеевые» мостики; 5 —«захваченная» вода
При содержании в исходной воде грубодисперсных и коллоидных веществ свыше 100 мг/кг коагуляция проводится в осветлителях, а при меньшей концентрации экономичнее ее организовать непосредственно на насыпных фильтрах. В этом случае имеют дело с прямоточной или контактной коагуляцией. При контактной коагуляции процесс отделения хлопьев происходит в фильтрующем слое. Контактная коагуляция представляет особый случай, когда мелкие частицы удерживаются на поверхности крупных зерен слоя. Она отличается большей скоростью протекания, процесса и почти полным извлечением из воды мелких частиц.
Для интенсификации процесса коагуляции часто в обрабатываемую воду вводят специальные вещества — флокулянты.
Сущность процесса флокуляции состоит в том, что агрегация коллоидных частиц в этом случае происходит не только непосредственно, но и через молекулы флокулянта. В качестве флокулянтов используются неорганические или органические высокомолекулярные соединения: активная кремнекислота, полиакриламид и др. Так, молекула полиакриламида диссоциирует и по кислотному, и по основному типу в зависимости от рН. В изоэлектрическом состоянии степень диссоциации полиакриламида по обоим типам одинакова. Однако несмотря на наличие у молекулы полиакриламида одновременно положительно и отрицательно заряженных ио-ногенных групп в целом она электронейтральна. Ионогенные группы молекул полиакриламида сорбируют различные частицы, образуя крупные структурированные системы. Следует заметить, что флокуляция не заменяет процесс коагуляции, а лишь углубляет и интенсифицирует его.
При коагуляции снижается бикарбонатная щелочность воды. Однако эквивалентно этому в воду при введении коагулянта попадают и ионы SO42- В то же время концентрации ионов Са2+ и Mg2+ при коагуляции не изменяются и происходит лишь замена карбонатной жесткости на некарбонатную.
Большое влияние на процесс коагуляции оказывает температура. При повышении температуры увеличиваются скорость и глубина гидролиза, скорость формирования и отделения твердой фазы, что в конечном случае ускоряет и углубляет коагуляцию примесей. Оптимальной для коагуляции воды сернокислым алюминием считается температура около 303-308 К.
При осуществлении технологического процесса очень большое значение имеет кинетика коагуляции. Практика показывает, что для создания оптимальных условий процесса необходимо сначала быстрое перемешивание воды с коагулянтом (<10мин), а затем процесс должен происходить в более спокойной гидродинамической обстановке.
В общем случае скорость коагуляции определяется количеством слипающихся частиц в единицу времени, причем различают медленную коагуляцию, когда не каждое столкновение 
 частиц приводит к их слипанию, и быструю, когда каждое столкновение оканчивается слипанием.
частиц приводит к их слипанию, и быструю, когда каждое столкновение оканчивается слипанием.
По теории М. Смолуховского скорость быстрой коагуляции в неподвижной воде описывается уравнением реакции второго порядка [2 ]
dn/dτ = k(no-n)2, (1)
где п0 — число частиц (счетная концентрация) в начале процесса;
п — число образовавшихся агрегатов; к —константа коагуляции.
Режим потока воды оказывает большое влияние на формирование хлопьев и, с учетом их непрочности, может даже способствовать разрушению сформировавшихся хлопьев. Поэтому скорость воды в зоне их формирования и отстаивания должна быть не более 1 —1,5 мм/с.
Если вводимые в воду сульфаты при коагуляции сернокислым алюминием оказывают благоприятное влияние на процесс коагуляции и частично выводятся вместе с осадком, то при коагуляции сернокислым железом они практически все попадают на следующую ступень очистки (ионообменную) и создают дополнительные трудности при их удалении из воды. Поэтому наиболее рационально вводить в обрабатываемую воду ионы этих металлов без анионов, затрудняющих в дальнейшем процессы обессоливания.
Этого можно достичь лишь при применении электрохимического метода (электрокоагуляции), основанного на анодном растворении металла в воде при прохождении через воду электрического тока. При этом очистка воды от коллоидных веществ осуществляется в ряде одновременно протекающих процессов: электрохимического растворения электродов с переходом ионов металла в раствор, окислительно-восстановительных реакций на электродах, собственно коагуляции, явлений электрофореза (движения частиц под влиянием внешнего электрического поля).
В основе электрокоагуляции лежит процесс анодного растворения металлов под действием постоянного электрического тока с последующим гидролизом катионов металлов и их участием в процессе коагуляции примесей воды.
Электрохимическое растворение металлов, погруженных в раствор и находящихся под действием приложенного извне электрического потенциала, зависит от многих факторов, но в основном определяется процессами, происходящими на аноде и катоде.
На аноде происходит окисление металла с переходом его ионов в раствор:
А1-Зе →А13 +; (2)
Fe-2e→Fe2 +. (3)
Непрерывный выход ионов металла в раствор возможен лишь при условии отвода электронов с катода, который осуществляется при протекании реакций восстановления.
В кислой среде (рН<7) электроны отводятся с катода по реакции
2H3O++2e→2H2++2OН-. (4)
В нейтральных и щелочных растворах электроны непосредственно ассимилируются молекулами воды с последующим образованием водорода и гидроксильных ионов:
2Н2О + 2е→Н2 + 2ОН-. (5)
При наличии в воде кислорода последний участвует в процессе ассимиляции электронов по реакции
2Н2О + О2 + 4е→4ОН-. (6)
Кислород также участвует в процессе перехода ионов Fe2 + в форму Fe3+ и при достаточно высоком потенциале выделяется на аноде, пассивируя металл (образуя пленки оксида). При этом вследствие поляризации электрода резко сокращается скорость его растворения. Поляризация катода происходит за счет отложений на его поверхности щелочноземельных соединений вследствие высоких значений рН воды в прикатодной области.
Ионы ОН- образуют с ионами металла гидроокиси: А1(0Н)3 или Fe(OH)2. Гидрозакись железа затем окисляется в гидроокись:
4Fe(OH)2 + O2 + H2O→4Fe(OH)3. (7)
Таким образом, гидроокиси металлов могут быть получены в воде без введения в нее анионов SO42-. Кроме того, электрохимический метод электрокоагуляция позволяет отказаться от использования традиционных коагулянтов, а также аппаратуры, связанной с их приготовлением и дозированием.
Теоретически количество электричества, необходимого для растворения 1 г-экв металла, составляет 26,8 А -г. Однако практически количество электричества всегда выше теоретического вследствие поляризационных эффектов на пластинах, расхода энергии на нагрев воды и т. п. Для уменьшения этого эффекта и более равномерного использования пластин производят через определенное время (обычно через 15 мин) переполюсовку подводимого напряжения. При проведении электрокоагуляции наблюдается повышение рН воды на 0,5— 1,0 за счет разрядки ионов Н +, что дает возможность не подщелачивать воду реагентами.
Практически во всех аппаратах для электрокоагуляции, применяемых в промышленности, используются пластины металла, что повышает стоимость обработки и увеличивает габариты аппарата (рисунок). Все конструкции пластинчатых аппаратов относятся к безнапорному типу. 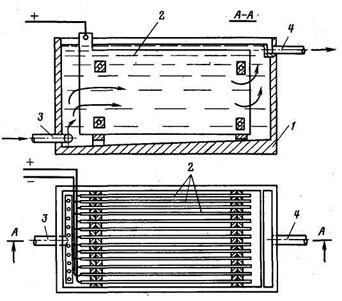
Рисунок - Пластинчатый электрокоагулятор:
1-корпус; 2-пластины из металла; 3-поступление воды; 4-выход воды.
При работе с большими расходами воды такие аппараты неудобны, их сложно располагать в схемах ВПУ. Применение пластинчатых аппаратов невыгодно также и потому, что использование пластин возможно только до 50%-ного их растворения. Иначе вследствие коробления пластин возможно короткое замыкание в аппарате.
Существует два способа подключения электродов к источнику питания: монополярный, когда одноименные пластины (например, аноды) подключаются к одному из полюсов источника питания, а катоды — к другому, и биполярный, когда питание подводится лишь к крайним электродам и общее падение напряжения на аппарате складывается из суммы падений напряжения на отдельных ячейках.
При биполярном подключении электродов требуется большее напряжение на аппарат, но меньшее количество контактов. При этом уменьшаются размеры аппарата и сечение подводящих кабелей, что упрощает монтаж и эксплуатацию электрокоагуляторов и уменьшает их стоимость.
Рекомендуемое напряжение на ячейках при расстоянии между электродами 10—12 мм для анодного растворения железа составляет 3 В, а для растворения алюминия—4В. Из опыта освоения электрокоагуляторов рекомендуется поддерживать плотность тока- около 10 А/м, а скорость потока
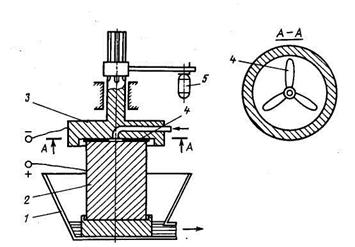
Рисунок - Электрокоагулятор с обновляемой поверхностью анода:
1 — сборная ванна; 2 — растворимый анод; 3 — катод; 4 — пластинки из абразивного материала; 5 — сервопривод; → — очищенная вода
воды между пластинами — не менее 0,5 м/с. Расход электроэнергии на электрокоагуляцию составляет 0,05—0,5 кВт-ч/т воды.

Рисунок - Электрокоагулятор типа «труба в трубе» с пористой засыпкой:
1— корпус; 2 — внутренняя труба-катод; 3 —вставной анод; 4 —сетка; 5—гранулы металла; 6 —вход воды; 7—выход воды
Эффективность электрокоагуляционной очистки воды существенно выше эффективности реагентной коагуляции и составляет 70—90% против 50—60% при реагентной коагуляции.
|
|
|
|
Дата добавления: 2014-01-13; Просмотров: 2837; Нарушение авторских прав?; Мы поможем в написании вашей работы!