КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Форма подготовки - очная 3 страница
|
|
|
|
Теории находятся в тесной связи и взаимозависимости. Они отражают разные стороны химической организации вещества, ее статику и динамику и дополняют друг друга. Структурные теории более известны (точнее их эволюция). Коснемся гносеологических корней появления кинетических теорий. Родоначальником этих теорий можно считать Вильгельми, который в 1850 году дал закон скорости химической реакции. Однако его работа (как и у Бертолле) оказалась непонятой большинством химиков, преждевременной. Исследование “законченных предметов” еще не подготовило почвы для систематического изучения процессов. Структурные теории, несмотря на их прогрессивность, рассматривали вещество в нереакционном состоянии. Вильгельми же впервые рассмотрел вещество в реакционной среде.
Следующим этапом в кинетических теориях стало представление о химическом равновесии. Возникла химическая статика, которая связала химию с термодинамикой. Статика, несмотря на ограниченность, вступила в противоречие с законом постоянства состава, кратных отношений, эквивалентов. Закон равновесия, закон действия масс в лучшем случае замалчивались, в худшем встречались в штыки. Только в 1861 году Менделеев ввел понятие об обратимости и равновесии в своей книге “Органическая химия” на примере синтеза и омыляемости сложных эфиров. Только с работами Гульберта и Вааге пришло признание закона действия масс и т.д. Химическая статика основывалась на господствовавших идеях механицизма (перенос из физики), поэтому она легче пробила себе путь, чем другие области кинетики, в частности, химическая динамика. В химии, как и в механике, наиболее естественным методом является рассмотрение равновесия сил притяжения и отталкивания.
|
|
|
Лишь во второй половине 1860х годов статика стала сменяться кинетической динамикой, как непрерывное изменение сил взаимодействия. Если вещества А и В соединяются с образованием продуктов реакции, то сила взаимодействия пропорциональна реагирующим массам веществ А и В:
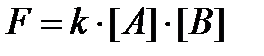
Отсюда один шаг до вывода о том, что нет принципиальной разницы между силами сродства в химических соединениях постоянного и переменного состава. 1870е–80е годы характеризуются тем, что идеи химического динамизма начинают овладевать умами химиков и становятся более заметными. Однако развитие идей химического динамизма пошло в двух направлениях. Одни сводили это к всеобщей подвижности, механическому перемешиванию тел, т.е. к отсутствию покоя атомов и молекул, что привело к известным осцилляционным и планетарным моделям, отказу от структурных представлений. Это развивали Бертло, Кеккуле, частично Менделеев. Меншуткин по этому поводу говорил, что едва ли теории строения, не обнимающие динамические стороны химических явлений, могут считаться выражением состояния химии.
Были и другие взгляды на динамизм: есть определенная взаимозависимость между атомами соответствующих частей, т.е. химическое соединение не есть что-то мертвое, застывшее, одаренное постоянным движением. Но свойства динамизма не рассматривались как функции химического строения. Фактически это было рассмотрение химического взаимодействия в нереагирующей молекуле.
Эти противоречия в динамизме не антагонистические, а дуалистические. Разрешаются при переходе от внереакционных систем к исследованию химического процесса (качества химического вещества). По Гульберту и Вааге реакции совершаются е вполне, но отчасти, они всегда подвижны и обратимы в зависимости от условий. Динамизм объясняется только лабильностью сродства, т.е. через непрерывное изменение энергии химического взаимодействия. Попытки свести динамизм к осцилляции и планетарным моделям является вульгарным динамизмом, который есть подновленный механицизм. Естественно, химическая динамика наиболее полное развитие получила в трудах Ван-Гоффа “Очерки по химической динамике” (1894). Академик Семенов оценил эту книгу как веху для развития человеческого знания. В ней дано понятие молекулярности реакции. Это не феноменологическая (понятийная) теория, но позволяющая исследовать механические процессы на начальном уровне. Примеров нормального участия целочисленного количества молекул в элементарном акте превращения, вопреки ожиданиям, немного. Идет описание аномальных процессов (“возмущающих действие”). Тогда это не все поняли. Было дано понятие стеночного катализа, автокатализа (продукты реакции – катализаторы), рекуперации энергии реакции (возвращения энергии от стадии к стадии). Это был первый труд, где сформулирована необходимость рассмотрения перехода от нереакционных к реакционным системам, т.е. учет участия в реакции всего-всего-всего.
|
|
|
Бутлеров в своей теории видел зачатки динамизма. Критически оценивая свою теорию, он не ограничивался представлениями о структуре, как о чем-то неподвижном. Ввел динамический фактор, таутомерию. Считал, что структурные теории отойдут на второй план после динамических, более прогрессивных теорий.
Одних теорий строения недостаточно для описания химического явления. Типичный пример – реакция Фаворского (1935), где есть переход протона, который играет в реакции определяющее значение. Эта реакция имеет каталитическую природу и проходит при неполновалентном взаимодействии реагирующих веществ с каталитической системой. Ведущую роль играет здесь не дискретность, а непрерывность связей при действии спиртового раствора щелочи. В данном случае химии молекул оказалось недостаточно для объяснения механизма процесса. В реакции Дэвиса-Измайлова происходит примерно то же самое. Оказалось, что таких примеров множество и все эти реакции идут в растворе, где происходит постоянное расслабление и перераспределение химических связей, и в концентрированных системах, а также в некоторых фазовых реакциях при взаимодействии со стенками сосудов. Во всех этих реакциях наряду с молекулами реагента принимают участие на правах сореагентов всякие молекулярные комплексы растворителей, примеси, катализаторы, ингибиторы и т.п., хотя их химизм заключается во взаимодействии протона и атома водорода реагентов, что является перекрыванием волновых функций. Часто для описания простых реакций недостаточно традиционных представлений химии молекул, а требуется привлечение реакционных систем.
|
|
|
Аргументы основных положений Ван-Гоффа переросли в цепную теорию Боденштейна-Семенова сначала для газовых и гомогенных процессов, потом и для гетерогенного катализа. Это синтез и дискретности с непрерывностью и структурных теорий. Дискретность наблюдается в дискретной валентной передаче от одной частицы к другой (свободный радикал). Непрерывность в том, что сам неспаренный электрон взаимодействует с радикалом, и это зависит от структуры радикала.
В чем же различие структурных и кинетических теорий? В отражении объективно существующего единства вещества. Кинетические теории отрешились от старых взглядов на незыблемость стехиометрических законов и теорию валентности как фундамент химии. Это было в работах по кинетике как наличие дробных порядков реакции. Цепная теория Боденштейна-Семенова продолжила это.
Дальнейшее развитие цепных теорий пошло по пути рассмотрения участия стенки сосуда в захвате и генерации свободных радикалов. Далее предметом теорий стал гетерогенный катализ. Его теория стала наглядным примером диалектического совмещения дискретного характера свободной валентности и непрерывной формы химического взаимодействия между атомами частицы и кристалла с образованием слабой, а затем и прочной хемосорбции.
Определяющую роль в химических процессах играет не столько электронное строение молекулярных реагентов, сколько электронное строение образующихся из них свободных радикалов, т.е. взаимодействие свободных электронов с остальными связями в сложном радикале. Именно в этом Семенов видит путь глубокого понимания синтеза Бутлеровской и кинетической теорий. Этот вывод принципиален, т.к. реакционная способность связана с электронной структурной особенностью индивидуальной молекулы. (даже сейчас).
|
|
|
Фактически в кинетических теориях произошел синтез структурных и динамических теорий. Они снимают противоречия дискретности непрерывности. По схеме состав – структура – свойства – процесс переходим к теориям химического процесса.
Лекция №6. (15 декабря 1998)
Химическая эволюция.
Проблема ХЭ раньше находилась на задворках. Генезис не был задачей для химика. Если рассматривать различные аспекты ХЭ, то окажется, что они важны как с познавательной, так и с методологической точки зрения. Этой проблемой интересуются биохимики, космохимики, геохимики, биологи и др. ХЭ определяют как происхождение и прогрессивное развитие химической формы движения материи как некой целостности или химической организации вещества во всех ее проявлениях. Значение ХЭ еще в том, что через нее можно пройти по иерархии теорий и подойти к эволюционным теориям катализа, а это связано, прежде всего, с самоорганизацией в неживых и живых системах.
Каковы аспекты ХЭ интересны? Геохимический аспект отображает историю химизма Земли. Хотя и не совпадает с теорией химической организации вещества, имеет право на существование. Он связан с появлением химической формы организации веществ из дохимической (перекликается с космохимией). По геохимическим представлениям существовал горячий материал – протопланеты, которые остывали, появлялись твердые вещества, далее шло отделение твердых фаз от жидких, образовывались химические формы. Т.е. от ионизированной плазмы дело доходило до высших форм.
По некоторым другим представлениям была, напротив, холодная масса, на которой под действием радиации шло развитие от простых к сложным радикалам, разделение твердых и жидких фаз, а дальше как в предыдущем описании.
По Вернадскому геохимия – история атомов на Земле, т.е. наука об истории планеты.
| Биологические формы Живое вещество (верхняя граница хим. формы дв-я материи, предбиологические системы) Адсорбционные слои, к-е упорядочивают систему, увеличивают концентрацию в-в Появление водных растворов (коллоидных растворов, в том числе органических в-в) Появление твердых кристаллических тел и осадочных пород Твердые оксидные и силикатные системы и расплавы – начало химической эволюции по земному типу Замороженные радикалы Ионизирующие плазмы (нижняя граница) |
Развитие форм химической организации вещества.
В условиях ионизирующей плазмы (10000К) возможно существование водорода, азота. При 5000К могут образовываться оксиды титана, циркония и т.д. Ниже 5000К появляются твердые частицы. При 3000К около 12 соединений могут быть в твердой фазе в виде карбидов гафния, тантала и т.д. При 1000К около 100 веществ могут становиться твердыми. Есть предположения, что жизнь зародилась во внеземных условиях, т.к. аминокислоты находят в метеорных телах.
Важна роль появления твердых тел в качестве подложек для образования растворов и расплавов. Это косвенный показатель роли бертоллидных систем в каталитических взаимодействиях бертоллидов и дальтонидов. Дальнейшее развитие шло, основываясь на водных системах.
Биогеохимический аспект развивался Вернадским. Биосфера – особая форма организации вещества, которая при взаимодействии с другими сферами участвует в биохимической эволюции Земли (состав, структура). Роль биогеохимии заключается в том, что дается общая направленность, но не затрагивается иерархия химической формы организации вещества (сделано на основе термодинамического аспекта).
Биохимический аспект. Биохимия со своей спецификой – единственный аспект, где проблема химической эволюции решается как таковая во всей ее полноте – от вопросов происхождения элементов, отбора структур до образования живого вещества. Хочется эволюцию свести к чистой биохимии. Общее между ней и химией – изучение химических процессов, общих закономерностей химизма. Различие состоит в том, что в химии рассматриваются все формы организации вещества как самостоятельные системы, а в биохимии – только высокоорганизованные формы, более сложные процессы, относящиеся к биологическим объектам. Биохимия не интересуется реакционной способностью объектов (что главное для химии), изучая только биоформу движения материи (но не с химических позиций). Тем не менее, биохимия и химия обогащают друг друга за счет разных подходов. Биохимический аспект химической эволюции интересен потому, что рассматривается переход от системы к системе, от простых атомов до сложных структур, а во времени охватывается период в сотни миллионов лет. Если у химика центральный интерес лежит в генезисе вещества, то у биохимии – в биогенезе. Биохимия берет у химии готовые методы определения состава, структуры и т.д. и ими пользуется. Химические понятия используются для интерпретации реакций изолированных ферментов. Поскольку химия не знает принципов актуализма, использующихся в биологии, принципов эволюционных учений, то за счет этого она обогащается. Биохимический аспект дает химикам направленность ХЭ, раскрывает движение силы ХЭ, поскольку используются методы историзма, эволюционной биологии, что является мощнейшим инструментарием. Биохимия указывает отдельные этапы и стадии ХЭ.
Говоря о направленности ХЭ, следует рассмотреть общую направленность развития химических форм движения. Наиболее простой формой движения считается механическое перемещение, далее по сложности идут всевозможные поля (физическая форма), далее химическая форма движения, а наверху стоит жизнь (своеобразная форма движения). Обычно ХЭ призвана рассматривать не только химическую форму движения материи как звено в ряду, но и как развитие этой формы движения во всей ее целостности. Главный фактор – прогрессивность в развитии, биогенез. Принцип восхождения от простого к сложному (от менее организованных систем к более организованным) позволяет расставить химические системы в соответствии с историзмом, прогрессивностью в развитии.
Такие попытки классификации были, но оказались малозаметны. В 1911 году Курилов предлагал рассматривать все системы от формы первичной материи до протоплазмы, т.е. в качестве перехода от менее высокого к более высокому. Пример – образование более высокоорганизованного ионного кристалла хлорида натрия из менее организованных ионов натрия и хлора: Na+ + Cl- ® NaCl.
Существуют критерии, которые могут разделить системы по высоте организации. К одному из таких критериев относится термодинамический аспект. Но для него благоприятной ситуацией считается, если есть простейшие неорганические системы. В этом случае термодинамика со своими первым и вторым началами все объясняла. Но эта классическая термодинамика запрещает процессы с переходом к биогенезу. Если в этой термодинамике направленность процесса относится к состоянию равновесия, то в химической эволюции – к устойчивому неравновесию. По второму началу термодинамики все процессы в изолированных системах протекают в направлении возрастания энтропии (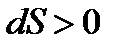 ). Отсюда идет закон неубывания энтропии, что определяет направленность процесса в сторону беспорядка, хаоса, а уменьшение энтропии может идти только в случае флуктуации (отклонения), что само по себе маловероятно, т.е. закон имеет вероятностный характер.
). Отсюда идет закон неубывания энтропии, что определяет направленность процесса в сторону беспорядка, хаоса, а уменьшение энтропии может идти только в случае флуктуации (отклонения), что само по себе маловероятно, т.е. закон имеет вероятностный характер.
В химической эволюции преобладает фактор устойчивого неравновесия, т.е. присутствует господство не вероятностных процессов. В данном случае нужна термодинамика открытых (неравновесных) систем, приводящая к понятию синергетики и т.п. Для того чтобы судить об открытых системах (в частности, о биосистемах) нужна такая термодинамика. Пригожин, Хакен получили нобелевскую премию в этой области за теорию диссипативных систем (почти чистая математика). По этой теории энтропия делится на составляющие: 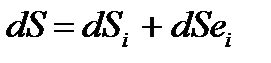 , где
, где 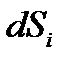 – прирост энтропии за счет внутренних энергетических процессов, а
– прирост энтропии за счет внутренних энергетических процессов, а 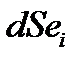 – учитывает изменение энтропии при обмене с окружающей средой. Этот подход устраняет запрет движения системы в сторону упорядочивания. Основным отличием живого от неживого (крайний случай) является то, что в живом возникает и воспроизводится низкоэнтропийная организация систем.
– учитывает изменение энтропии при обмене с окружающей средой. Этот подход устраняет запрет движения системы в сторону упорядочивания. Основным отличием живого от неживого (крайний случай) является то, что в живом возникает и воспроизводится низкоэнтропийная организация систем.
Николаев дает классификацию систем по степени их организации.
1. Механические системы, условием равновесия которых является равенство нулю виртуальной работы внешних сил, приложенных к системе. Описываются классической механикой, где нет понятия температуры как таковой.
2. Термодинамические системы, условием равновесия которых является максимум энтропии при минимуме свободной энергии. Описываются классической термодинамикой, которая не пользуется понятием времени, хотя в целом предназначена для описания процессов.
3. Открытые системы, которые появляются при взаимодействии систем второго типа. Эти системы являются потоками (в том числе потоками энтропии, диссипативные системы) и описываются термодинамикой необратимых процессов. Изменение энтропии по времени стремится к минимуму (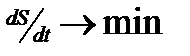 ). При
). При 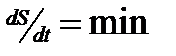 система достигает максимально устойчивого состояния или стационарного режима, поток становится равномерным. При выведении из этого состояния система стремится обратно. Это первый элемент самоорганизации вещества.
система достигает максимально устойчивого состояния или стационарного режима, поток становится равномерным. При выведении из этого состояния система стремится обратно. Это первый элемент самоорганизации вещества.
4. Живые биосистемы. Стабильность поддерживается саморегулированием.
Системы 3 и 4 являются высокими, возникающими из систем низшего порядка, когда те находятся в состоянии перехода к равновесию, но не в состоянии равновесия (флуктуации). Системы 3 и 4 более важны для химической эволюции и являются системами высшего уровня.
Примерами систем 3 ранга из химии могут служить автокаталитические системы, в которых регулирование скорости может идти в две стороны. Здесь есть потоки, связанные с переносом электронов, протонов, атомных групп. Будучи открытыми, эти системы с низкой энтропией как бы оберегают себя от хаоса, распада, развала посредством автокаталитических и ингибирующих механизмов с избытком производящих энтропию.
Появление систем 4 ранга с точки зрения химической эволюции происходит за счет усложнения составляющих частей системы и за счет усложнения механизмов связей таких систем. Основную роль играет не столько природа связей, сколько способность проявлять обратные связи, т.е. способность реагировать на внешние возбудители.
С таким термодинамическим подходом можно провести границу между живым и неживым. Неживое имеет тенденции к изменению неорганических систем 1 и 2 уровней, склонно к разупорядочиванию, рассеянию энергии, ликвидации флуктуаций, т.е. стремится к максимуму энтропии, к состоянию равновесия. У систем 3 и 4 ранга (живого) все наоборот – имеются тенденции к минимуму энтропии, к способности локального аккумулирования энергии химической реакции, к флуктуации, к нарушению равновесия, к устойчивому неравновесию. Простой пример таких систем – человек. Живой в состоянии устойчивого неравновесия, а мертвый пришел в равновесие.
Устойчивость систем 1 и 2 уровней обеспечивается изолированным характером от внешнего воздействия, а устойчивость систем 3 и 4 ранга обусловливается их открытым характером, обменом веществ с окружающей средой и механизмами обратной связи. Потоки – суть динамической устойчивости.
Существуют и другие подходы, связанные с философскими понятиями. Например, информационный подход, в соответствии с которым сложность вещества характеризуется информационным уровнем вещества (принесен Ждановым из кибернетики). Сущность подхода: чем выше химическая организация вещества, тем более чувствительным рецептором (датчиком) внешних воздействий оно становится. Воздействие механических, тепловых, электрических полей фиксируется в виде возбужденного состояния при сохранении своей индивидуальности. Все это связано с теорией отражения, где есть понятия отклика на воздействие с сохранением памяти (первоначального образа) этого материального объекта.
Если взять органическое вещество, то от неорганического вещества оно будет отличаться тем, что сохраняет свою индивидуальность. Может быть гидрофилом, гидрофобом, иметь полифункциональность, иметь способность аккумулировать энергию, чтобы войти в сопряженную реакцию. Подход рассчитывает информационную емкость вещества на один атом. Критерием повышения организации вещества здесь является повышение динамичности, способность к комплиментарности (взаимное соответствие структур), к кодированию, к дистанционному возбуждению и обратной связи. Количественной мерой информационной емкости является способность к отражению.
Биологическая организация вещества.
У биологов есть понятия отбора, актуализации, принципа преобразования развития. Эти вещи помогают химику понять, как в химической эволюции шел отбор химических элементов, структур, подход к предбиологическим формам, которые имеют признаки биообъектов.
Более системный биоподход в ХЭ.
Высокоорганизованные химические системы – это динамически устойчивые системы, состояния устойчивого неравновесия. Они характеризуются лабильностью (здесь: подвижностью связей, перестройкой вещества).
По Бауэру первой особенностью живого является процесс самодвижения системы, ее активность, которая состоит в противодействии внешним силам, активность против равновесия, несовместимого с жизнью. Принцип устойчивого неравновесия – это универсальный закон биологической формы движения материи. Чтобы система функционировала, нужно пополнять свои запасы структурной энергии, повышать уровень неравновесия, динамичности и непрерывной структурной лабильности за счет обмена веществом и энергией с внешней средой.
Поскольку рассматривается биоподход, то факторы из биологии можно перенести в химию. Отбор химических элементов (в истории, в ХЭ). В основу живого вошли органогены (азот, кислород, фосфор, сера и др.). Живое на 97% построено из них. В качестве активных центров ферментов вошли еще 12 элементов (натрий, кальций, калий), ионная сила раствора обеспечивается хлором. Они составляют около 1.6% в живом. Остальные 20 элементов, входящие в состав живого “весят” около 1%. Причем эти цифры не соответствуют распространенности этих элементов в природе (азота в сумме с углеродом менее 0.024%). Из 2млн. химических соединений (известных науке) около половины приходится на органические вещества, состоящие всего из 6–18 элементов. Здесь играют роль биохимические факторы – лабильность строения, динамическая устойчивость органогенов. Почему углерод лежит в основе? Этот элемент уникален, поскольку он и донор, и акцептор в связи, имеет различные степени окисления, образует циклы, цепи и т.п. Для органогенов характерно образование внутримолекулярных и внутрикомплексных связей, что обеспечивает все богатство химической связи. Способны имитировать связи, характеризующиеся полупроводниковыми свойствами (органические полупроводники), образовывать макроэргические связи, обладающие огромным запасом энергии (молекулы АТФ). Способны образовывать слабые водородные связи, определяющие лабильность, многоцентровые связи (пирольные кольца иминов). Если рассматривать ХЭ с точки зрения отбора, то элементов было отобрано немного, что обусловлено биохимическими факторами, лежащими в основе жизнедеятельности.
Отбор структур – вторая стадия, соответствующая в биологии естественному отбору. Из 2млн. в построении живого принимают участие только несколько сот, из ста аминокислот в белки входит около двадцати, лишь четыре нуклеотида участвуют в передаче наследственности. Из такого малого круга структур состоит весь живой и растительный мир!
После перехода к живому ХЭ застыла. Уже отобрались все структуры и элементы, способные поддерживать динамизм устойчивого неравновесия. В древних окаменелостях нашли те же соединения, что и в современных животных, т.е. ХЭ стала. И химику нужно понять, каким образом природа приспосабливает для своих нужд низкоорганизованные материалы (углекислый газ, воду и т.п.) и при обычных температурах и давлении ведет синтез сложнейших соединений из простых.
В ходе ХЭ отобрались структуры, которые обеспечили виды связи, регулирующие саму структуру. Как шла эволюция? Первыми появились фазовые границы (основа физико-химической адсорбции), что вело к концентрации и упорядочиванию вещества. Это первое проявление возможности каталитических эффектов. Вторыми образовывались полупроводниковые цепи, шел перенос протонов, трансгидрирование, появлялись группы окислительно-восстановительного и кислотно-щелочного катализа. Третий фрагмент эволюции включат образование групп, ответственных за энергетическое обеспечение структур – оксо-, оксигруппы, фосфорсодержащие ферменты с макроэргическими связями, которые также начинают выполнять роль катализаторов. Их главная роль состоит в снятии термодинамических запретов путем сопряжения реакции диспропорционирования макроэргических связей с ферментативными реакциями. На четвертом этапе развивались полимерные структуры РНК и ДНК, исполняющие роль шаблона или катализаторных матриц, которые могут воспроизвести себе подобные структуры – эффект подложки катализатора. Пятыми появились структуры со связями типа металл–азот, металл–углерод, как один из типов высокоорганизованных соединений.
Отношением к катализу объясняется отбор этих структур. Роль катализа высока – это ведущий фактор ХЭ, поскольку общее у всех пяти этапов – отношение структур к катализу, проявление каталитических функций.
На ранних стадиях ХЭ катализ был не нужен, т.к. энергии было в избытке (тепло, разряды, радиация и т.д.), перекрывающей энергетический барьер. При 5000К появлялись первые твердые частицы, космопыль. В этих условиях уже возможно проявление каталитической активности. При охлаждении эти частицы исполняли роль неспецифических катализаторов в газофазных реакциях, катализаторов роста кристаллообразующихся физических тел. Нужно отметить, что неорганические структуры дезактивируют каталитическую активность элементов, по сравнению с органическими. Например, каталитическая активность железа в минерале меньше активности свободного иона Fe+3, но при включении железа в порфириновое кольцо активность повышается на 10 порядков. Органическая ветвь катализаторов ведет к активации (является более высокой ступенью).
Наибольшие шансы эволюционировать имели структуры, способные к катализу. Есть мнения, что на ранних стадиях при образовании оксидов, сульфидов и других минералов (апатиты), те являлись матрицами для синтеза белка, т.к. структурно соответствуют протобелковым молекулам (Бернал, Костецкий).
Гомогенный катализ совершеннее гетерогенного. А если взять катализ металлорганическими соединениями, то это ступень перехода к высокоспецифичному катализу ферментами.
Таким образом, отбор структур шел по степени отношения к катализу. Высока роль бертоллидного состояния, т.к. соединения такого типа обладали набором активных центров с различной энергией связи, которые могли обеспечить взаимодействие с другим веществом в качестве катализатора с образованием молекул дальтонидов. Но есть различия в принципе действия химических и биокатализаторов. Последние представляют собой особого рода системы, промежуточные между бертоллидами и дальтонидами. Их бертоллидное качество состоит в широком спектре энергоемкости, зарядов связи, а дальтонидное – в строгом составе частиц с определенными активными энергетически локализованными центрами и в высокой стереоспецифичности. Другими словами биокатализаторы являются ключом для одного единственного субстрата, который реализуется на данном активном центре.
Лекция №7.
Продолжение раскрытия темы химической эволюции.
Интенсивно развивается система ферментоподобных катализаторов. Одни считают, что, изучая изолированный фермент, можно решить проблему. Другие полагают, что нужно изучать фермент in vivo – в среде обитания, а затем переносить in vitro. Другими словами, существует два подхода для изучения биокатализаторов.
В отличие от обычной химической биореакция протекает не путем увеличения энтропии, а посредством уменьшения энтропии переходных состояний, вследствие чего получаются более организованные системы по схеме фермент – субстрат. При увеличении же энтропии образуется менее организованная система (более хаотичная). В создании биокатализаторов нужно стремиться к воссозданию всей системы со всеми ее параметрами и ее поддержанию. Биокатализаторы работают в определенной системе и при образовании активных комплексов.
Пусть стоит задача создания стабильных каталитических систем. Нужно in vitro решить, затем удовлетворить in vivo. Это идет через познание процессов биогенеза эволюции, что решают химия и биология.
Взаимодействуя со средой, биокатализатор воспроизводит свой каталитический аппарат. Если в обычных системах 1 и 2 ранга преобладает направление от хаоса к равновесию, то в 3 и 4 – прогрессирующее развитие, что является не просто процессом (реакцией) обеспечивающим функционирование таких высокоорганизованных систем.
В 1847 году Берцелиус говорил, что с помощью катализаторов удастся подслушать тайну лаборатории живого организма, т.е. используя опыт живой природы, можно создать катализатор. Это же направление развивали Либих, Митчерлих, Густавсон и другие.
|
|
|
|
|
Дата добавления: 2014-12-08; Просмотров: 443; Нарушение авторских прав?; Мы поможем в написании вашей работы!