КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Загальна нозологія 7 страница
г) порушеннями використання ВЖК як джерела енергії (зменшення активності ферментів Р-окиснення і циклу Кребса, наприклад, при гіпоксії).
Для того щоб запобігти ушкоджувальній дії надлишку жирових кислот, клітина має у своєму розпорядженні систему ферментів, які переводять вільні жирові кис-
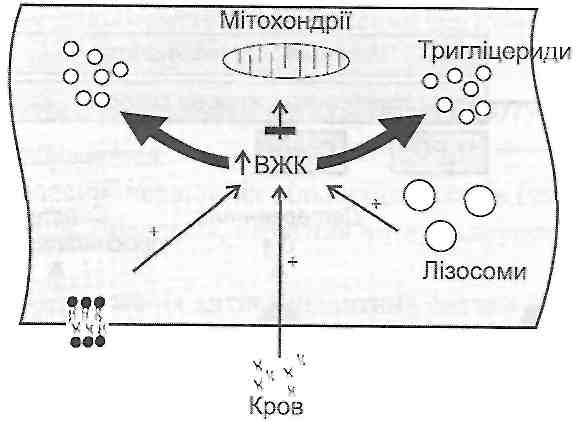
Рис. 35. Причини накопичення вільних жирових кислот (ВЖК) у клітині
лоти у форму триглщеридів. При цьому відбувається невластиве для норми відкладення тригліцеридів у вигляді жирових крапель, тобто виникає жирова дистрофія клітини.
11.14. У яких випадках іони кальцію втягуються в патогенез ушкодження клітини? З якими ефектами цих іонів пов 'язана їхня участь в ушкодженні клітинних структур?
Ушкодження клітинних структур може бути обумовлене стійким підвищенням концентрації іонів Са2+ у цитоплазмі клітини. Така ситуація виникає або в результаті надмірного надходження іонів Са2+ у цитоплазму (гіперкальціємія, підвищення проникності плазматичної мембрани), або в результаті порушення механізмів, що забезпечують видалення іонів Са2+ із цитоплазми (порушення Са-насосів, Na-Ca-об-мінного механізму, Са-акумулгоючої функції мітохондрій).
Підвищення концентрації іонів Са2т у цитоплазмі викликає:
а) контрактуру (пер є скорочення) фібрилярних структур клітини (міофібрил, елементів цитоскєлета);
б) активацію фосфоліпази А,;
в) роз'єднання процесів окиснення й фосфорування.
11. 15. Чим можуть бути обумовлені зміни вмісту іонів натрію й калію в клітині і яка роль таких порушень у патогенезі клітинного ушкодження?
Вирівнювання концентрацій іонів Na+ і К + по обидва боки плазматичної мембрани (збільшення вмісту Na+ і зменшення вмісту К+ у цитоплазмі) у своїй основі може мати два механізми: 1) посилену дифузію іонів через плазматичну мембрану за існуючим концентраційним і електричним градієнтом і 2) порушення механізмів активного транспорту Na+ і К+ (Na-K-насоса).
Перший механізм реалізується в умовах загальних порушень водно-електролітного обміну (гіпернатріємія, гіпокаліємія) і порушень бар'єрної функції плазматичної мембрани (підвищення її іонної проникності).
Розлади функції Na-K-насоса можуть бути обумовлені дефіцитом АТФ у клітині, збільшенням вмісту холестеролу в ліпідному бішарі мембрани (наприклад, при атеросклерозі), дією цілої низки специфічних інгібіторів Na-K-АТФ-ази (наприклад, строфантину).
Зміни вмісту іонів Na+ і К+ викликають:
а) втрату клітиною електричного мембранного потенціалу (потенціалу спокою), а отже і збудливості;
б) набряк клітини;
в) осмотичне розтягнення клітинних мембран, що супроводжується підвищенням їх проникності.
11. 16. Чим може бути обумовлений розвиток внутрішньоклітинного ацидозу і які зміни в клітині можуть бути з ним пов 'язані?
До розвитку внутрішньоклітинного ацидозу можуть спричинятися:
1) надмірне надходження іонів Н+ у клітину з позаклітинного середовища (декомпенсований газовий або негазовий ацидоз);
2) надмірне утворення кислих продуктів у самій клітині при активації гліколізу (молочна кислота), порушеннях циклу Кребса (три- і дикарбонові кислоти), гідролітичному розщепленні фосфоліпідів клітинних мембран (вільні жирові кислоти, фосфорна кислота) та ін.;
3) порушення зв'язування вільних іонів Н+ у результаті недостатності буферних систем клітини;
4) порушення виведення іонів Н+ з клітини при розладах Na-H-обмінного механізму, а також в умовах порушеного місцевого кровообігу в тканині.
Внутрішньоклітинний ацидоз викликає: а) зміну конформації білкових молекул з порушенням їх ферментативних, скоротливих та інших властивостей; б) підвищення проникності клітинних мембран; в) активацію лізосомних гідролітичних ферментів.
11.17. Які зміни білкових молекул мають значення в патогенезі ушкодження клітини?
До білкових (протеїнових) механізмів ушкодження клітини можна віднести:
1) інгібуванння ферментів (оборотне і необоротне);
2) денатурацію, тобто порушення нативної будови білкових молекул у результаті обумовлених розривом ковалентних зв'язків змін вторинної й третинної структур білка;
3) протеоліз, що здійснюється під дією лізосомних протеолітичних ферментів (ка-тепсинів) і протеаз, які активуються іонами Са2+, У результаті протеолізу можуть з'являтися пептиди, що мають властивості фізіологічно активних речовин. З виходом останніх з ушкоджених клітин може бути пов'язаний розвиток як місцевих, так і загальних реакцій організму (запалення, гарячка).
11. 18. Які порушення функціонування генетичного апарату клітини можуть призводити до її ушкодження?
Основу ушкодження клітини можуть становити так звані нуклеїнові механізми, обумовлені порушеннями процесів:
1) реплікації ДНК (денатурація ДНК, ушкодження ДНК-репліказної ферментної системи, дефіцит трифосфонуклеотидів - АТФ, ГТФ, ТТФ і ЦТФ);
2) транскрипції (мутаційні дефекти генної матриці, інгібування ДНК-залежної РНК-полімерази антибіотиками й токсинами, порушення посттранскрипційної модифікації інформаційної РНК: неприєднання "кепа" до головного кінця молекули, порушення утворення полі-А-хвоста, розлади сплайсингу тощо);
3) трансляції (дефіцит або якісні зміни інформаційної, рибосомної або транспортної РНК, а також рибосомних ферментів і нєферментних білків; дефіцит вільних амінокислот і АТФ; інгібування процесу антибіотиками й мікробними токсинами).
11.19. Які існують універсальні механізми підвищення проникності клітинних мембран при ушкодженні клітини?
Підвищення проникності клітинних мембран може бути обумовлене:
1) активацією пероксидного окиснення ліпідів;
2) активацією фосфоліпаз;
3) осмотичним розтягненням мембран;
4) адсорбцією білків (поліелектролітів) на мембрані;
5) змінами фазового стану мембранних ліпідів (ацидоз, зміна температури).
/ 1.20. Які порушення виникають у клітині в результаті ушкодження окремих її органоїдів (плазматичної мембрани, мітохондрій, ендоплазматичногоретикулуму лізосом)?
Порушення бар'єрної функції плазматичної мембрани призводить до вирівнювання існуючих у нормі концентраційних градієнтів речовин: у клітину надходять іони Na+, Са2+, СІ", а виходять іони К+, Mg2+, неорганічного фосфату, низько- і високо-молекулярні органічні сполуки (АМФ, АДФ, проміжні продукти клітинного обміну, білки-ферменти). З ушкодженнями білків і глікопротеїдних комплексів, вбудованих у плазматичну мембрану, пов'язані порушення систем активного транспорту речовин (Na-K-, Са-насосів; Na-Ca- і Na-H-обмінних механізмів); зміни специфічних іонних каналів (Na-, K-, Са-каналів); порушення клітинних рецепторів, що сприймають зовнішні регуляторні сигнали (а- і)3-адренорецепторів, ш- і n-холінорецепторів та ін.); порушення міжклітинних взаємодій; зміни антигенних властивостей клітини.
Ушкодження мітохондрій супроводжується або пригніченням процесів клітинного дихання, або ефектом роз'єднання процесів окиснення й фосфорування. І в тому, і в другому випадку результатом розладів мітохондріальних функцій буде порушення енергозабезпечення клітини (рис. 36).
Ушкодження шорсткого ендоплазматичного ретикулуму призводить до деза-грегації полісом, унаслідок чого порушуються реакції біосинтезу білка в клітині. У результаті ушкодження гладкого ендоплазматичного ретикулуму і його ферментних систем страждають процеси детоксикації, мікросомного окиснення та ін. У деяких
клітинах, наприклад м'язових, порушується здатність ендоплазматичного (саркоплазма-тичного) ретикулуму депонувати іони Са2+, що сприяє реалізації так званих кальцієвих механізмів ушкодження клітини.
Підвищення проникності лізосомних мембран призводить до виходу в цитоплазму гідролітичних ферментів, активація яких у кінцевому підсумку викликає необоротні зміни клітини — її аутоліз.
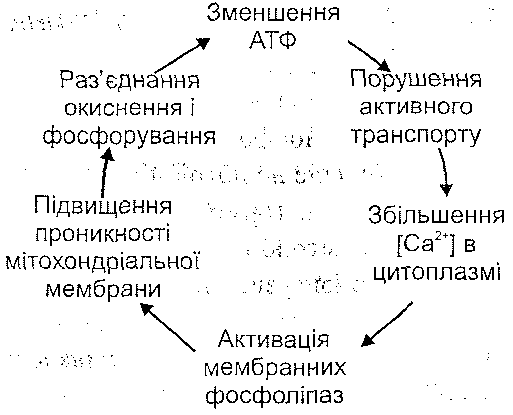
Рис. 36. Роль дефіциту АТФ в ушкодженні клітини
11.21. Які існують механізми загибелі клітин? Чим вони принципово відрізняються?
Розрізняють два механізми загибелі клітин: некроз і апоптоз.
Некроз - це загибель необоротно ушкоджених клітин, яка відбувається за участю лізосомних ферментів, що здійснюють гідролітичне розщеплення усіх компонентів клітини (аутоліз), руйнують плазматичну мембрану і зумовлюють вихід вмісту клітини за ЇЇ межі в навкружну тканину з наступним розвитком запалення.
Апоптоз— це шлях запрограмованої загибелі клітин ("самогубство"), що відбувається за участю спеціально призначених для цього активованих ферментів (кас-паз), які руйнують власну ДНК, ядерні білки і білки цитоскелета, не ушкоджуючи при цьому плазматичну мембрану.
Основні відмінності між некрозом і апоптозом представлено в табл.
| Властивості | Некроз | Апоптоз |
| Об'єм клітини | Збільшений (набряк) | Зменшений (стиснення) |
| Ядро | Пікноз → каріорексис —f каріолізис | Фрагментація - розпад на окремі фрагменти (нуклеосоми) |
| Плазматична мембрана | Розірвана | Інтактна |
| Вміст цитоплазми | Ферментативне перетравлювання лізосомними ферментами (аутоліз), вихід вмісту клітини за її межі | Інтактний |
| Викликає запалення в тканині | Часто | Ніколи |
| Фізіологічне чи патологічне явище | Завжди патологічне (кульмінація незворотного ушкодження клітин) | Часто фізіологічне (генетично запрограмована загибель клітин); може бути патологічним після деяких видів ушкодження клітин, особливо після ушкодження ДНК |
11.22. Назвіть основні причини апоптозу.
I. Фізіологічний апоптоз відбувається:
а) у період ембріогенезу (імплантація, органогенез, онтогенетична інволюція, метаморфоз) відповідно до генетичної програми знищення клітин, які вже більше не потрібні;
б) під час гормон-залежної інволюції органів у дорослих (загибель клітин ен-дометрію під час менструального циклу, атрезія фолікулів яєчника під час менопаузи, зменшення маси молочних залоз після припинення лактації);
в) у популяціях клітин з високою інтенсивністю проліферативних процесів для підтримання сталої кількості клітин (наприклад, у криптах слизової тонкої кишки);
г) у клітинах, що вже виконали свою функцію (у нейтрофілах по завершенню гострого запалення, у лімфоцитах у кінці імунної відповіді;
ґ) у потенційно небезпечних для організму лімфоцитах (клітинах так званих "заборонених клонів'1);
д) під час дії цитотоксичних Т-лімфоцитів (Т-кілерів) на уражені вірусами і пухлинні клітини.
II. Апоптоз в патологічних умовах може відбуватися:
а) унаслідок дії патогенних чинників, що спричиняють необоротне ушкодження ДНК (іонізуюча радіація, протипухлинні препарати);
б) в інфікованих вірусами клітинах (наприклад, при вірусному гепатиті);
в) у паренхіматозних органах у процесі розвитку атрофії, що виникає як наслідок обтурації їхніх вивідних проток (підшлункова і слинні залози);
г) у пухлинах — як під час їхньої регресії, так і в процесі активного росту;
ґ) під впливом чинників, що збільшують проникність мітохондріальних мембран.
11.23. Як здійснюється апоптоз?
Розрізняють три послідовні фази апоптозу. І. Фаза ініціювання. Суть її полягає в послідовній активації так званих "суїцид- них" ферментів — каспаз (їх сьогодні налічують понад 10). Існує два шляхи такої активації: зовнішній і внутрішній.
Зовнішній (рецепторопосередкований) шлях пов'язаний з існуванням на поверхні клітинної мембрани так званих "рецепторів смерті" (із сімейства рецепторів до фактора некрозу пухлин - ФНП). Взаємодія цих рецепторів з ФНП та деякими іншими лігандами спричиняється до появи в цитоплазмі (з внутрішнього боку плазматичної мембрани поблизу активованого рецептора) б ілка-п о середника, який активує протеазу- каспазу-8 (у людини- каспазу-10). Активна каспаза-8 започатковує каскад реакцій (у тому числі і аутокаталітичних), під час яких відбувається перетворення неактивних прокаспаз в активні каспази - ферменти, що "вбивають" клітину.
Внутрішній (мітохондріальний) шлях пов'язаний зі збільшенням проникності мітохондріальних мембран (утворенням "мітохондріальної пори") та виходом із мітохондрій у цитоплазму так званих проапоптичних сполук- цитохрому с,
апоптозіндукуючого фактора (АІФ) та ін. Утворення "мітохондріальної пори " відбувається внаслідок заміни антиапоптичних білків в мембрані мітохондрій на проапоптичні білки. Така заміна має місце тоді, коли припиняється дія на клітину факторів росту та інших стимуляторів клітинної активності. Цитохром с, потрапивши в цитоплазму, разом з білком цитозолю - апоптозактивуючим фактором (ААФ) утворює комплекс, який активує каспазу-9. Зазначена протеаза започатковує процес утворення інших активних каспаз (див. вище).
II. Фаза вбивання ("екзекуції"). У результаті наведених вище подій утворюються активні форми каспаз-екзекуторів (каспаза-3, каспаза-6 та ін.). Ці протеази: а) розщеплюють білки цитоскелета; б) руйнують білки матриксу ядра; в) активують цитоплазматичну ДНК-азу через розщеплення її білкового інгібітора. Унаслідок цього змінюються форма і об'єм клітини, ядро розпадається на окремі фрагменти, а сама клітина на так звані апоптичні тільця — утвори, що зовні мають мембрану, а всередині містять спресовані органели і окремі фрагменти ядра.
Ш. Фаза вилучення загиблих клітин. Здійснюється макрофагами шляхом фагоцитозу мертвих клітин і апоптичних тілець. Цьому сприяє вивільнення клітинами ще на ранніх фазах їхнього апоптозу речовин-хемотаксинів, а також поява на поверхні клітин, що гинуть, білків-маркерів, які дають змогу макрофагам розпізнавати мертві клітини серед ще живих. Важливе значення цього етапу полягає в тому, що своєчасний фагоцитоз загиблих клітин запобігає їхньому некрозу, виходу з клітин лізосомних ферментів і розвитку запалення.
11.24. Які захисно-компенсаторні механізми має ушкоджена клітина?
Все різноманіття захисно-компенсаторних реакцій клітини у відповідь на її ушкодження можна умовно розділити на дві групи.
I. Реакції, спрямовані на відновлення порушеного внутрішньоклітинного гомеостазу:
а) активація механізмів активного транспорту речовин (Na-K-, Са-насосів; Na-Ca-, Na-H-обмінних механізмів, мікровезикулярного транспорту);
б) посилення регенерації антиоксидантів;
в) зв'язування вільних жирових кислот (синтез тригліцеридів);
г) активація синтезу білків, нуклеїнових кислот, фосфоліпідів та ін. Неодмінною умовою реалізації цих механізмів є достатнє енергозабезпечення
клітини. Це досягається підвищенням інтенсивності енергетичного обміну (активація гліколізу, клітинного дихання, пентозного циклу) і перерозподілом наявних у клітин енергетичних ресурсів.
II. Реакції, спрямовані на створення функціонального спокою ушкодженої клітини. їхня мета полягає в тому, щоб усунути можливі додаткові зрушення внутрішньоклітинного гомеостазу при дії фізіологічних збуджувальних факторів (стабілізація ушкодження) і звести до мінімуму енергетичні витрати на виконання специфічних функцій клітини.
До таких реакцій можна віднести:
а) утворення клітиною простагландинів і блокада ними |3-адренорецепторів (рис. 37);
б) інгібування адеі-іілатциклази і підвищення активності фосфодіестерази, що руйнує цАМФ;
в) утворення аденозину - природного блокатора Са-каналів та ін.

Рис. 37. Захисна роль простагландинів при ушкодженні клітини
11.25. Які існують підходи до патогенетичного лікування ушкоджених клітин?
Основні принципи впливу на ушкоджені клітини:
1) обмеження і пригнічення молекулярних механізмів ушкодження (блокада Са-каналів, застосування антиоксидантів, інгібіторів фосфоліпази А2 і протеаз, активація біосинтезу білків та ін.);
2) створення функціонального спокою (щадний режим, дієта, блокада клітинних рецепторів та ін.);
3) енергетичне й пластичне забезпечення гомеостатичних механізмів клітини (вплив на периферичний кровообіг і мікроциркуляцію з метою поліпшення доставки кисню і поживних речовин до ушкоджених клітин, введення енергетичних і пластичних субстратів).
12. Екстремальні стани
12.1. Що таке екстремальні стани?
Екстремальні стани - це стани організму, що характеризуються надмірною напругою або виснаженням пристосувальних механізмів. Екстремальні стани можуть розвиватися первинно при дії на організм різноманітних надзвичайних подразників (наприклад, травм, екзогенних інтоксикацій, різких коливань температури повітря й концентрації кисню) або стати результатом несприятливого перебігу наявного захворювання (наприклад, недостатності кровообігу, дихальної, ниркової або печінкової недостатності, анемії та ін.).
Найбільш важливими екстремальними станами і такими, що найчастіше зустрічаються, є шок, колапс, кома.
12.2. Що таке шок?
Шок - це важкий патологічний процес, що супроводжується виснаженням життєво важливих функцій організму і приводить його на межу життя і смерті через критичне зменшення капілярного кровообігу в уражених органах.
12.3. Назвіть основні види шоку.
Залежно від причин виникнення виділяють такі види шоку: травматичний, геморагічний (див. розд. 26), опіковий (див. розд. 5), турнікетний (розвивається після зняття джгута через чотири години й більше після його накладення), ангідремічний (див. розд. 23), кардіогенний (див. розд. 27), панкреатичний (див. розд. ЗО), анафілактичний (див. розд. 10), септичний, інфекційно-токсичний та ін.
Залежно від первинних механізмів, що лежать в основі патогенезу шоку, виділяють:
1) гіповолемічний шок (геморагічний, ангідремічний);
2) шок, пов 'язаний з порушеннями насосної функції серця (кардіогенний);
3) судинні форми шоку (анафілактичний, панкреатичний);
4) больовий шок, при якому порушується центральна регуляція кровообігу (травматичний, опіковий).
12.4. Які механізми лежать в основі порушень загальної гемодинаміки і мікроциркуляції при шоку?
Незалежно від причин виникнення шок виявляє себе комплексом порушень гемодинаміки, для якого характерні зменшення артеріального тиску, хвилинного об'єму серця, венозного повернення до серця, об'єму циркулюючої крові, об'ємної швидкості органного кровообігу; порушення реологічних властивостей крові (агрегація формених елементів, підвищення в'язкості крові). Комплекс цих порушень може бути визначений як гостра недостатність кровообігу.
Відповідно до законів гемодинаміки первинне порушення одних її показників при будь-якому різновиді шоку веде вторинно до порушень усіх інших.
В основі розвитку розладів кровообігу за умов шоку можуть лежати такі механізми.
I. Зменшення об'єму циркулюючої крові:
1) крововтрата (геморагічний шок);
2) втрата плазми крові при великому ексудативному запаленні (опіковий шок);
3) вихід рідини з кровоносних судин у тканини при генералізованому підвищенні проникності судин (анафілактичний шок);
4) зневоднення (ангІдремічний шок);
5) перерозподіл крові в судинному руслі (тромбоз і емболія магістральних вен).
II. Зменшення хвилинного об'єму серця:
1) порушення скоротливої функції серця (інфаркт міокарда);
2) тампонада серця (розрив серця, ексудативний перикардит);
3) аритмії (фібриляція шлуночків).
III. Зменшення загального периферичного опору - генерал із оване розширення судин:
1) падіння нейрогенного тонусу артеріол (больові форми шоку);
2) зменшення базального тонусу судин під дією біологічно активних речовин (анафілактичний, панкреатичний шок) або токсичних продуктів (травматичний, турнікетний, інфекційно-токсичний шок).
IV. Порушення реологічних властивостей крові:
1) синдром внутрішньо судинного дисемінованого зсідання крові (панкреатичний шок);
2) агрегація формених елементів крові (септичний, інфекційно-токсичний шок);
3) згущення крові - гемоконцентрація (ангІдремічний шок).
12.5. Чим визначаються важкі наслідки шоку?
Розлади загальної гемодинаміки і мікроциркуляції, що виникають за умов шоку, небезпечні насамперед порушеннями кровообігу: а) мозкового; б) вінцевого; в) ниркового. Результатом зазначених розладів є прогресуюче порушення центральної регуляції життєво важливих функцій, аж до розвитку коми; виникнення явищ гострої серцево-судинної і ниркової недостатності. Патогенні фактори, що виникають при цьому, — гіпоксія, ацидоз та інтоксикація - ведуть до генералізованого і незворотного ушкодження клітин.
12.6. Поясніть патогенез травматичного шоку.
Травматичний шок розвивається внаслідок великих за обсягом ушкоджень тканин. У його клініці розрізняють дві стадії: збудження (еректильну) і гальмування (торпідну).
Стадія збудження короткотривала, для неї характерний стан збудження центральної нервової системи, що виникає в результаті надходження великої кількості больових імпульсів з ушкоджених тканин. Розвивається больовий стрес, що виявляє себе посиленням функцій системи кровообігу, дихання, деяких ендокринних залоз
(аденогіпофіза, мозкової і кіркової речовини надниркових залоз, нейросекреторних ядер гіпоталамуса) з вивільненням у кров великої кількості кортикотропіну, адреналіну, норадреналіну, вазопресину (див. розд. 33).
Стадія гальмування більш тривала (від кількох годин до доби) і характеризується розвитком у центральній нервовій системі гальмівних процесів. Генера-лізоване гальмування захоплює й центри життєво важливих функцій (кровообігу, дихання), вони порушуються, унаслідок чого розвивається кисневе голодування. Гіпоксія, у свою чергу, збільшує порушення в серцево-судинному й дихальному центрах. Розлади гемодинаміки і зовнішнього дихання прогресують - "зачароване коло "замикається.
Крім нервово-рефлекторних механізмів у виникненні і розвитку травматичного шоку певну роль відіграє токсемія, обумовлена всмоктуванням у кров продуктів розпаду нежиттєздатних тканин. Особливе значення надають так званому ішемічному токсину.
Участь токсичних продуктів у патогенезі травматичного шоку доводиться дослідами з "перехресним кровообігом", коли після відтворення шоку в однієї тварини явища цього патологічного процесу виникають у другої, інтактної, пов'язаної з першою загальним кровообігом.
12.7. Що таке колапс?
Колапс — це судинна недостатність, що швидко розвивається і характеризується в першу чергу падінням судинного тонусу, а також гострим зменшенням об'єму циркулюючої крові. При цьому відбувається зменшення припливу венозної крові до серця, зниження серцевого виштовху, падіння артеріального й венозного тиску, порушуються перфузія тканин і обмін речовин, настає гіпоксія головного мозку, пригнічуються життєво важливі функції організму.
12.8. Що таке краш-синдром?
Краш-синдром (синдром тривалого роздавлювання) - це патологічний процес, який розвивається у потерпілих у результаті тривалого (4-8 г і більше) роздавлювання м'яких тканин кінцівок уламками зруйнованих будинків, споруд, брилами ґрунту при обвалах у шахтах та ін.
У перебігу краш-синдрому розрізняють три періоди: 1) ранній (до 3-х діб) з переважанням явищ шоку; [ 2) проміжний (з 3-ї до 12-ї доби) з переважанням гострої ниркової недостатності; 3) пізній (з 8-12-ї доби до 1-2 міс), або період видужання, з переважанням місцевих симптомів.
У розвитку краш-синдрому велике значення мають три патогенетичних фактори:
а) больове подразнення;
б) травматична токсемія, обумовлена всмоктуванням токсичних продуктів аутолі-зу тканин з вогнища ураження;
в) плазмо- і крововтрата, пов'язані з набряком і крововиливами в зоні роздавлених або довготривало ішемізованих тканин.
12.9. Що таке кома?
Кома— це патологічний стан, що характеризується глибоким пригніченням функцій центральної нервової системи і виявляє себе втратою свідомості, відсутністю рефлексів на зовнішні подразники і розладами регуляції життєво важливих функцій організму.
12.10. Як класифікують коматозні стани?
Залежно від етіології розрізняють екзогенні й ендогенні коми.
1. Екзогенні коми виникають унаслідок дії патогенних факторів зовнішнього середовища або в результаті дефіциту факторів, необхідних для існування організму. Прикладами екзогенних ком є травматична (виникає при ушкодженні головного мозку), гіпоксична, гІпер- і гіпотермічна, екзотоксична, аліментарно-дистрофічна (виникає при важкому голодуванні).
2. Ендогенні коми розвиваються внаслідок порушень діяльності функціональних систем організму. Так, при розладах функції системи кровообігу може виникати апоплексична кома, системи крові — гемолітична, видільної системи - уремічна, печінки — печінкова, ендокринної системи — діабетична, гіпоглікемічна, гіпокор-тикоїдна, тиреотоксична та інші види коми.
12.11. Які механізми лежать в основі патогенезу коматозних станів?
1. Енергодефіцитний механізм. Порушення синтезу АТФ у нейронах центральної нервової системи призводить до генералізованого порушення їхніх функцій. Зазначений механізм є провідним у розвитку гіпоксичної, апоплексичної, респіраторної, гемолітичної, гіпоглікемічної, аліментарно-дистрофічної, екзотоксичної ком.
2. Порушення синаптичної передачі в центральній нервовій системі. Вони можуть бути пов'язані з: а) порушенням синтезу, транспорту, депонування і секреції нейромедіаторів; б) витісненням нейромедіаторів так званими псевдомедіаторами (хибними медіаторами); в) надмірною активацією гальмівних постсинаптичних рецепторів; г) блокадою збудливих постсинаптичних рецепторів. Зазначений механізм відіграє велику роль у розвитку печінкової, уремічної і екзотоксичної ком.
3. Загальні водно-електролітні порушення. Мають значення зміни осмотичного тиску крові й порушення балансу електролітів в організмі. Цей механізм є провідним у розвитку гіперосмолярної діабетичної коми, гіпертермічної, гіпокортикоїд-ної та гіпопаратиреоїдної ком.
4. Порушення кислотно-основного стану. Лежать в основі виникнення ацидотич-ної діабетичної, лактацидемічної, хлоргідропенічної та деяких інших ком.
5. Підвищення внутрішньочерепного тиску. Цей механізм діє при первинних ураженнях головного мозку. Він є основним у патогенезі коми при менінгітах та енцефалітах, а також травматичної коми при черепно-мозкових травмах.
13. Місцеві розлади кровообігу Порушення мікроциркуляції
13.1. Назвіть основні форми місцевих розладів кровообігу.
Артеріальна гіперемія, венозна гіперемія, ішемія, стаз, тромбоз, емболія.
13.2. Що таке артеріальна гіперемія?
Артеріальна гіперемія - це збільшення кровонаповнення органа або тканини за рахунок надмірного надходження крові по артеріальних судинах.
13.3. Які функціональні зміни й клінічні ознаки характеризують артеріальну гіперемію?
При артеріальній гіперемії спостерігають розширення дрібних артерій, артеріол, вен і капілярів; прискорення течії крові в них, пульсацію дрібних артерій і капілярів, збільшення числа видимих оком судин, збільшення тиску в артеріолах, капілярах і венах. У результаті зазначених змін виникає почервоніння, підвищується місцева температура, збільшується об'єм гіперемованої ділянки, підвищується тургор тканини, посилюються обмін речовин і функція органа.
73.4. Які фактори можуть бути причиною артеріально)' гіперемії? Що мають на увазі, коли кажуть про фізіологічну й патологічну артеріальну гіперемію?
Причиною артеріальної гіперемії може бути вплив фізичних, хімічних і біологічних факторів зовнішнього середовища; збільшення навантаження на орган або ділянку тканини; психогенні впливи.
ФЬіологічною називають артеріальну гіперемію, що виникає під дією звичайних фізіологічних подразників (збільшення навантаження на орган, психогенні впливи). Основними її різновидами є робоча й реактивна гіперемія.
Робоча гіперемія - це збільшення течії крові в органі під час посилення його функції (збільшення вінцевого кровообігу при посиленні роботи серця, гіперемія слинних залоз під час приймання їжі та ін.).
Реактивна гіперемія являє собою збільшення течії крові після його короткочасного обмеження. Розвивається зазвичай у нирках, головному мозку, шкірі, кишках, м'язах.
|
|
Дата добавления: 2014-12-07; Просмотров: 381; Нарушение авторских прав?; Мы поможем в написании вашей работы!