КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Скорость химических реакций 2 страница
|
|
|
|
Метод валентных связей (МВС) window.top.document.title = "3.3.5. Метод валентных связей (МВС)";
Метод валентных связей (МВС) иначе называют теорией локализованных электронных пар, поскольку в основе метода лежит предположение, что химическая связь между двумя атомами осуществляется с помощью одной или нескольких электронных пар, которые локализованы преимущественно между ними. В отличие от ММО, в котором простейшая химическая связь может быть как двух-, так и многоцентровой, в МВС она всегда двухэлектронная и обязательно двухцентровая. Число элементарных химических связей, которые способен образовывать атом или ион, равно его валентности. Так же, как и в ММО, в образовании химической связи принимают участие валентные электроны. Волновая функция, описывающая состояние электронов, образующих связь, называется локализованной орбиталью (ЛО).
Отметим, что электроны, описываемые ЛО, в соответствии с принципом Паули должны иметь противоположно направленные спины, то есть в МВС все спины спарены, и все молекулы должны быть диамагнитны. Следовательно, МВС принципиально не может объяснить магнитные свойства молекул.
Тем не менее, принцип локализованных связей имеет ряд важных преимуществ, одно из которых – его чрезвычайная наглядность. МВС достаточно хорошо, например, предсказывает валентные возможности атомов и геометрию образующейся молекулы. Последнее обстоятельство связано с так называемой гибридизацией АО. Она была введена для объяснения того факта, что двухэлектронные двухцентровые химические связи, образованные за счет АО в разных энергетических состояниях, имеют одинаковую энергию. Так, Be*(2s11p1), B*(2s12p2), C*(2s12p3) образуют за счет s- и p-орбиталей соответственно две, три и четыре связи, а потому одна из них должна быть прочнее других. Однако опыт показывает, что в BeH2, BCl3, CH4 все связи равноценны. У BeH2 угол связи равен 180°, у BCl3 – 120°, а у CH4 – 109°28'.
|
|
|
Согласно представлению о гибридизации, химические связи образуются смешанными – гибридными орбиталями (ГО), которые представляют собой линейную комбинацию АО данного атома (s- и p-АО Be, B, C), обладают одинаковыми энергией и формой, определенной ориентацией в пространстве (симметрией). Так s- и p-орбитали дают две sp-ГО, расположенные под углом 180° друг относительно друга.
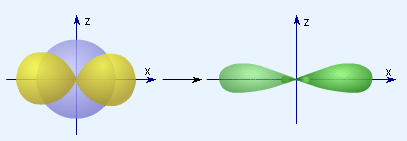
|
| s-орбиталь + p-орбиталь и две sp-ГО. |
В молекуле CH4 гибридные орбитали из четырех АО углерода (одной s и трех p), называются sp3-орбиталями, они полностью эквивалентны энергетически и пространственно направлены к вершинам тетраэдра.
Таким образом, когда один атом образует несколько связей, а его валентные электроны принадлежат разным орбиталям (s и p; s, p и d), для объяснения геометрии молекул в МВС необходимо привлекать теорию гибридизации атомных орбиталей. Основные положения теории следующие:
1. Введение гибридных орбиталей служит для описания направленных локализованных связей. Гибридные орбитали обеспечивают максимальное перекрывание АО в направлении локализованных σ-связей.
2. Число гибридных орбиталей равно числу АО, участвующих в гибридизации.
3. Гибридизуются близкие по энергии валентные АО независимо от того, заполнены они в атоме полностью, наполовину или пусты.
4. В гибридизации участвуют АО, имеющие общие признаки симметрии.
Согласно табл. 3.3 гибридные орбитали дают молекулы с углами 180°, 120°, 109°28', 90°. Это правильные геометрические фигуры. Такие молекулы образуются, когда все периферические атомы в многоэлектронной молекуле (или ионе) одинаковы и их число совпадает с числом гибридных орбиталей. Однако, если число гибридных орбиталей больше числа связанных атомов, то часть гибридных орбиталей заселена электронными парами, не участвующими в образовании связи, – несвязывающими или неподеленными электронными парами.
|
|
|
|
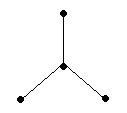
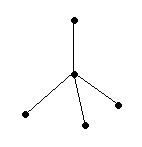
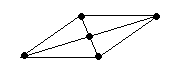
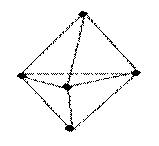
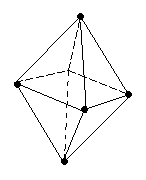
В качестве примера рассмотрим молекулы NH3 и H2O. Атомы азота и кислорода склонны к sp3-гибридизации. У азота на sp3-ГО, поимо трех связывающих пар электронов, образующих связь с тремя атомами водорода, остается одна несвязывающая пара. Именно она, занимая одну sp3-ГО, искажает угол связи H–N–H до 107,3°. В молекуле H2O таких несвязывающих пар две, и угол H–O–H равен 104,5° (рис.).
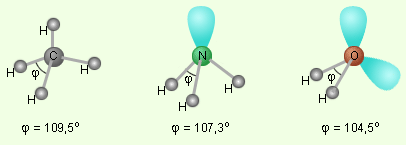
|
| Несвязывающие электронные пары и углы связи в молекулах NH3 и H2O в сравнении с молекулой CH4. |
Электроны связывающих и несвязывающих пар по-разному взаимодействуют между собой. Чем сильнее межэлектронное отталкивание, тем больше условный объем, занимаемый электронной парой. Для качественного объяснения экспериментальных фактов обычно считается, что несвязывающие пары занимают больший объем, чем связывающие, а объем связывающих пар тем меньше, чем больше электроотрицательности периферийных атомов (метод Гиллеспи).
Сравнительная характеристика ММО и МВС window.top.document.title = "3.3.6. Сравнительная характеристика ММО и МВС";
Оба квантовомеханических подхода к описанию химической связи – ММО и МВС – приближенны, ММО придает преувеличенное значение делокализации электрона в молекуле и основывается на одноэлектронных волновых функциях – молекулярных орбиталях. МВС преувеличивает роль локализации электронной плотности и основывается на том, что элементарная связь осуществляется только парой электронов между двумя атомами.
Сравнивая МВС м ММО, следует отметить, что достоинством первого является его наглядность: насыщаемость связи объясняется как максимальная ковалентность, направленность вытекает из направленности атомных и гибридных орбиталей; дипольный момент молекулы складывается из дипольных моментов связей, разности ОЭО атомов, образующих молекулу, и наличия неподеленных электронных пар.
|
|
|
Однако существование некоторых соединений невозможно объяснить с позиций МВС. Это электронодефицитные соединения (B2H6, NO,  ) и соединения благородных газов. Их строение легко объясняет ММО. Устойчивость молекулярных ионов и атомов в сравнении с молекулами легко предсказывается с позиции ММО. И, наконец, магнетизм и окраска вещества также легко объясняются ММО.
) и соединения благородных газов. Их строение легко объясняет ММО. Устойчивость молекулярных ионов и атомов в сравнении с молекулами легко предсказывается с позиции ММО. И, наконец, магнетизм и окраска вещества также легко объясняются ММО.
Количественные расчеты в ММО, несмотря на свою громоздкость, все же гораздо проще, чем в МВС. Поэтому в настоящее время в квантовой химии МВС почти не применяется. В то же время качественно выводы МВС гораздо нагляднее и шире используются экспериментаторами, чем ММО. Основанием для этого служит тот факт, что реально в молекуле вероятность пребывания данного электрона между связанными атомами гораздо больше, чем на других атомах, хотя и там она не равна нулю. В конечном счете, выбор метода определяется объектом исследования и поставленной задачей.
Введение в термодинамику window.top.document.title = "4.1. Введение в термодинамику";
Химические реакции сопровождаются выделением или поглощением энергии. Если энергия выделяется или поглощается в виде теплоты, то такие реакции записываются посредством уравнений химической реакций с указанием тепловых эффектов, при этом необходимо указывать фазовый состав реагирующих веществ.
Химические реакции, протекающие с выделением тепла, называются экзотермическими, а с поглощением тепла – эндотермическими. Изучением тепловых эффектов реакций занимается термохимия. В термохимии тепловой эффект реакции обозначается Q и выражается в кДж. Термохимия составляет один из разделов химической термодинамики, изучающей переходы энергии из одной формы в другие и от одной совокупности тел к другим, а также возможность, направление и глубину осуществления химических и фазовых процессов в данных условиях. Каждое отдельное вещество или их совокупность представляет собой термодинамическую систему. Если термодинамическая система не обменивается с окружающей средой ни веществом, ни энергией, ее называют изолированной. Такая идеализированная система используется как физическая абстракция при рассмотрении процессов, исключающих влияние внешней среды. Система, обменивающаяся с окружающей средой только энергией, называется закрытой. Если же возможен энергетический и материальный обмен – система открытая.
|
|
|
Состояние системы определяется термодинамическими параметрами состояния – температурой, давлением, концентрацией, объемом и т. д. Система характеризуется, кроме того, такими свойствами как внутренняя энергия U, энтальпия H, энтропия S, энергия Гиббса G. Из изменение в ходе химических реакций характеризуют ее энергетику системы. Перечисленные свойства системы зависят от температуры, давления, концентрации, поэтому они называются функциями состояния, не зависят от пути процесса и определяются только конечным и начальным состояниеми системы.
Внутренняя энергия системы U складывается из энергии движения и взаимодействия молекул, энергии связи в молекулах, энергии движения и взаимодействия электронов и ядер и т. п.
Абсолютная величина внутренней энергии не может быть определена, но ее изменение при переходе системы из начального состояния в конечное в результате осуществления химического процесса поддается расчету. Если система получает некоторое количество тепла при постоянном давлении Qp, последнее расходуется на изменение внутренней энергии системы ΔU и совершение работы A = PΔV против внешних сил:
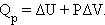
Это уравнение выражает закон сохранения энергии или первое начало термодинамики.
Адиабатический процесс – это процесс квазистатического расширения или сжатия газа в сосуде с теплонепроницаемыми стенками. Первый закон термодинамики для адиабатического процесса принимает вид A = –ΔU.
Изотермический процесс – это процесс квазистатического расширения или сжатия вещества, находящегося в контакте с тепловым резервуаром, (T = const).
Так как внутренняя энергия идеального газа зависит только от температуры (закон Джоуля), то первый закон термодинамики для изотермического процесса записывается в виде: Q = A.
При изохорическом процессе (V = const) поглощение или выделение тепла (тепловой эффект) связано только с изменением внутренней энергии: 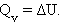
В химии чаще всего рассматривают изобарические процессы (P = const), и тепловой эффект в этом случае называют изменением энтальпии системы или энтальпией процесса: 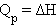
ΔH = ΔU + PΔV
Энтальпия window.top.document.title = "4.2. Энтальпия";
Энтальпия системы (от греч. enthalpo нагреваю), однозначная функция Н состояния термодинамической системы при независимых параметрах энтропии S и давлении P, связана с внутренней энергией U соотношением Н = U + PV
где V – объем системы.
В химии чаще всего рассматривают изобарические процессы (P = const), и тепловой эффект в этом случае называют изменением энтальпии системы или энтальпией процесса:
ΔH = ΔU + PΔV
Энтальпия имеет размерность энергии (кДж). Ее величина пропорциональна количеству вещества; энтальпия единицы количества вещества (моль) измеряется в кДж∙моль–1.
В термодинамической системе выделяющуюся теплоту химического процесса условились считать отрицательной (экзотермический процесс, ΔH < 0), а поглощение системой теплоты соответствует эндотермическому процессу, ΔH > 0.
Уравнения химических реакций с указанием энтальпии процесса называют термохимическими. Численные значения энтальпии ΔH указывают через запятую в кДж и относят ко всей реакции с учетом стехиометрических коэффициентов всех реагирующих веществ. Поскольку реагирующие вещества могут находиться в разных агрегатных состояниях, то оно указывается нижним правым индексом в скобках: (т) – твердое, (к) – кристаллическое, (ж) – жидкое, (г) – газообразное, (р) – растворенное. Например, при взаимодействии газообразных H2 и Cl2 образуются два моля газообразного HCl. Термохимическое уравнение записывается так: 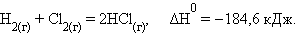
При взаимодействии газообразных H2 и O2 образующаяся H2O может находиться в трех агрегатных состояниях, что скажется на изменении энтальпии: 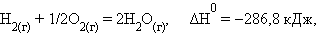
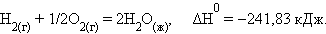 Приведенные энтальпии образования (реакций) отнесены у стандартным условиям температуры и давления (T = 298 K, P = 101,325 кПа). Стандартное состояние термодинамической функции, например, энтальпии, обозначается нижним и верхним индексами: ∆Н0298. Нижний индекс обычно опускают: ΔH0.
Приведенные энтальпии образования (реакций) отнесены у стандартным условиям температуры и давления (T = 298 K, P = 101,325 кПа). Стандартное состояние термодинамической функции, например, энтальпии, обозначается нижним и верхним индексами: ∆Н0298. Нижний индекс обычно опускают: ΔH0.
Введение в термохимию window.top.document.title = "4.3. Введение в термохимию";
Стандартная энтальпия образования ∆Н0обр – тепловой эффект реакции образования одного моля вещества из простых веществ, его составляющих, находящихся в устойчивых стандартных состояниях.
Например, для реакций
| Реакция | Энтальпия образования |
| Na2O(т) + H2O(ж) = 2NaOH(т) | 
|
| 1/2Na2O(т) + 1/2H2O(ж) = NaOH(т) | 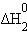
|
| Na(т) + 1/2O2(г) + 1/2H2(г) = NaOH(т) | 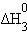
|
| 2Na(т) + O2(г) + H2(г)= 2NaOH(т) | 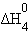
|
только∆Н0з является стандартной энтальпией образования NaOH.
Энтальпия образования простых веществ принята равной нулю, причем нулевое значение энтальпии образования относится к агрегатному состоянию, устойчивому при T = 298 K. Так, для йода 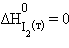 кДж∙моль–1,
кДж∙моль–1, 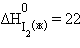 кДж∙моль–1,
кДж∙моль–1, 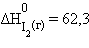 кДж∙моль–1. Для углерода
кДж∙моль–1. Для углерода  = 0 кДж∙моль–1,
= 0 кДж∙моль–1, 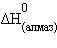 = 1,83 кДж∙моль–1.
= 1,83 кДж∙моль–1.
Стандартная энтальпия сгорания  – тепловой эффект реакции сгорания одного моля вещества до образования высших оксидов. Для органических веществ – до CO2(г) и H2O(ж). Теплота сгорания негорючих веществ принимается равной нулю. Теплота сгорания топлива характеризует его теплотворную способность.
– тепловой эффект реакции сгорания одного моля вещества до образования высших оксидов. Для органических веществ – до CO2(г) и H2O(ж). Теплота сгорания негорючих веществ принимается равной нулю. Теплота сгорания топлива характеризует его теплотворную способность.
Энтальпия растворения складывается из теплоты разрушения кристаллической решетки (ΔHреш > 0) и теплоты гидратации (сольвататции для неводных растворов), выделяющейся в результате взаимодействия молекул растворителя с молекулами или ионами растворяемого вещества с образованием соединений переменного состава – гидратов (сольватов) (ΔHгидр < 0).
В зависимости от соотношения значений ΔHреш и ΔHгидр энтальпия растворения может иметь как положительное, так и отрицательное значение.
Так, энтальпия растворения КОН – отрицательная величина и характеризует экзотермический процесс:

Растворение же KNO3(к) – эндотермический процесс (ΔH = 35,9 кДж∙моль–1), так как на разрушение кристаллической решетки (ΔH0реш = 684,5 кДж∙моль–1) затрачивается больше энергии, чем выделяется при гидратации ионов K+ и 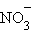 : –339 и –309,6 кДж∙моль–1 соответственно.
: –339 и –309,6 кДж∙моль–1 соответственно.
Стандартная энтальпия нейтрализации ΔH0нейтр– энтальпия реакции взаимодействия сильных кислот и оснований с образованием одного моля H2O при стандартных условиях.
| HCl + NaOH = NaCl + H2O; |
| H+ + OH– = H2O, ΔH0 = –55,9 кДж∙моль–1. |
Для концентрированных растворов сильных электролитов ΔH0нейтр может быть различным из-за изменения значения ΔHгидр их ионов при разбавлении.
Стандартная энтальпия реакции ΔH0 – тепловой эффект реакции определенного числа молей реагентов, задаваемого уравнением реакции при стандартных условиях. Например, для реакции
| 4H2O(ж) + 2Fe(т) → Fe2O3(т) + 4H2(г), ΔH0 = 321,3 кДж |
ΔH0 относится целиком к реакции, как она записана.
Стандартная энтальпия разрыва связи ΔH0св (называемая также энергией связи Eсв) – энергия, поглощаемая при разрыве связей двух атомов одного моля вещества, находящегося в газообразном состоянии при 298 К:
| HCl(г) → H(г) + Cl(г), ΔH0 = 429,7 кДж. |
Средние стандартные энтальпии связи могут быть определены для индивидуального соединения или путем усреднения значений, найденных для целых классов соединений.
Закон Гесса window.top.document.title = "4.4. Закон Гесса";
Пользуясь табличными значениями ΔH0обр, ΔH0сгор и Ecв, можно рассчитать энтальпии различных химических процессов и фазовых превращений. Основанием для таких расчетов является закон Гесса, сформулированный петербургским профессором Г. И. Гессом (1841 г.): «Тепловой эффект (энтальпия) процесса зависит только от начального и конечного состояния и не зависит от пути перехода его из одного состояния в другое».
Анализ закона Гесса позволяет сформулировать следующие следствия:
1. Энтальпия реакции равна разности сумм энтальпий образования конечных и начальных участников реакций с учетом их стехиометрических коэффициентов.
| ΔH = ΣΔHобр.конечн – ΣΔHобр.нач |
2. Энтальпия реакции равна разности сумм энтальпий сгорания начальных и конечных реагентов с учетом их стехиометрических коэффициентов.
| ΔH = ΣΔHсгор.нач – ΣΔHсгор.конечн |
3. Энтальпия реакции равна разности сумм энергий связей Eсв исходных и конечных реагентов с учетом их стехиометрических коэффициентов.
В ходе химической реакции энергия затрачивается на разрушение связей в исходных веществах (ΣEисх) и выделяется при образованиии продуктов реакции (–ΣEпрод). Отсюда
| ΔH0 = ΣEисх – ΣEпрод |
Следовательно, экзотермический эффект реакции свидетельствует о том, что образуются соединения с более прочными связями, чем исходные. В случае эндотермической реакции, наоборот, прочнее исходные вещества.
При определении энтальпии реакции по энергиям связей уравнение реакции пишут с помощью структурных формул для удобства определения числа и характера связей.
4. Энтальпия реакции образования вещества равна энтальпии реакции разложения его до исходных веществ с обратным знаком.
| ΔHобр = –ΔHразл |
5. Энтальпия гидратации равна разности энтальпий растворения безводной соли 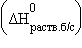 и кристаллогидрата
и кристаллогидрата 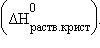
Из вышесказанного видно, что закон Гесса позволяет обращаться с термохимическими уравнениями как с алгебраическими, т. е. складывать и вычитать их, если термодинамические функции относятся к одинаковым условиям.
Например, диоксид углерода можно получить прямым синтезом из простых веществ (I) или в две стадии через промежуточный продукт (II):
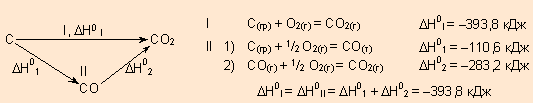
|
| Энтальпия первого пути равна сумме энтальпий отдельных стадий второго пути. |
Эти термохимические реакции можно представить в виде энтальпийных диаграмм. Естественно, за начало следует принять стандартные состояния простых веществ, энтальпии которых равны нулю. Образование сложных веществ (CO и CO2) сопровождается понижением энтальпии системы.
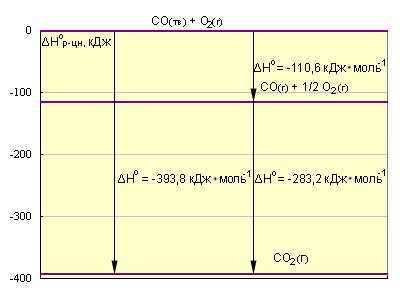
|
Энтропия window.top.document.title = "4.5. Энтропия";
Изменение энтальпии системы не может служить единственным критерием самопроизвольного осуществления химической реакции, поскольку многие эндотермические процессы протекают самопроизвольно. Иллюстрацией этого служит растворение некоторых солей (например, NH4NO4) в воде, сопровождающееся заметным охлаждением раствора. Необходимо учитывать еще один фактор, определяющий способность самопроизвольно переходить из более упорядоченного к менее упорядоченному (более хаотичному) состоянию.
Энтропия (S) – термодинамическая функция состояния, которая служит мерой беспорядка (неупорядоченности) системы. Возможность протекания эндотермических процессов обусловлена изменением энтропии, ибо в изолированных системах энтропия самопроизвольно протекающего процесса увеличивается ΔS > 0 (второй закон термодинамики).
Л. Больцман определил энтропию как термодинамическую вероятность состояния (беспорядок) системы W. Поскольку число частиц в системе велико (число Авогадро NA = 6,02∙1023), то энтропия пропорциональна натуральному логарифму термодинамической вероятности состояния системы W:
|
Размерность энтропии 1 моля вещества совпадает с размерностью газовой постоянной R и равна Дж∙моль–1∙K–1. Изменение энтропии *) в необратимых и обратимых процессах передается соотношениями ΔS > Q / T и ΔS = Q / T. Например, изменение энтропии плавления равно теплоте (энтальпии) плавления ΔSпл = ΔHпл/Tпл Для химической реакции изменение энтропии аналогично изменению энтальпии
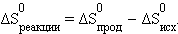
|
*) термин энтропия был введен Клаузиусом (1865 г.) через отношение Q/T (приведенное тепло).
Здесь ΔS0 соответствует энтропии стандартного состояния. Стандартные энтропии простых веществ не равны нулю. В отличие от других термодинамических функций энтропия идеально кристаллического тела при абсолютном нуле равна нулю (постулат Планка), поскольку W = 1.
Энтропия вещества или системы тел при определенной температуре является абсолютной величиной. В табл. ниже приведены стандартные энтропии S0 некоторых веществ.
| ||||||||||||||||||||||||||||||||||||||||||||
| Стандартные энтропии некоторых веществ. |
 (Дж∙моль–1∙K–1)
(Дж∙моль–1∙K–1)
Из табл. следует, что энтропия зависит от:
1. Агрегатного состояния вещества. Энтропия увеличивается при переходе от твердого к жидкому и особенно к газообразному состоянию (вода, лед, пар).
2. Изотопного состава (H2O и D2O).
3. Молекулярной массы однотипных соединений (CH4, C2H6, н-C4H10).
4. Строения молекулы (н-C4H10, изо-C4H10).
5. Кристаллической структуры (аллотропии) – алмаз, графит.
Наконец, рис. ниже иллюстрирует зависимость энтропии от температуры.
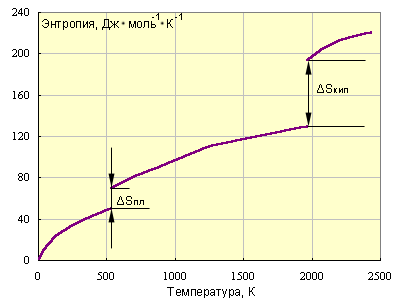
|
| Зависимость энтропии от температуры для свинца: ΔSпл = 8Дж∙моль–1∙K–1; Тпл = 600,5K; ΔSкип = 88Дж∙моль–1∙K–1; Tкип = 2013K. |
Следовательно, стремление системы к беспорядку проявляется тем больше, чем выше температура. Произведение изменения энтропии системы на температуру TΔS количественно оценивает эту тендецию и называется энтропийным фактором.
|
|
|
|
|
Дата добавления: 2014-12-07; Просмотров: 575; Нарушение авторских прав?; Мы поможем в написании вашей работы!