КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Наследственные ГА (эндоэритроцитарные)
Обусловлены генетическими нарушениями морфологии эритроцитов (мембранопатии), метаболизма этих клеток (энзимопатии) и синтеза гемоглобина (гемоглобинопатии).
Наследственные мембранопатии. Основным патогенетическим звеном ГА в результате мембранопатий являются наследственные изменения в первичной последовательности аминокислот одного из структурных мембранных белков эритроцитов (в частности, отсутствия белка спектрина) и дефекты липидных компонентов мембран этих клеток. Среди них наиболее часто встречается наследственный микросфероцитоз (болезнь Минковского - Шоффара), который наследуется по аутосомно-доминантному типу. Заболевание чаще развивается в юношеском, молодом и зрелом возрасте, хотя может возникать и у детей. Патогенез данного вида анемии представлен на схеме 23-1.
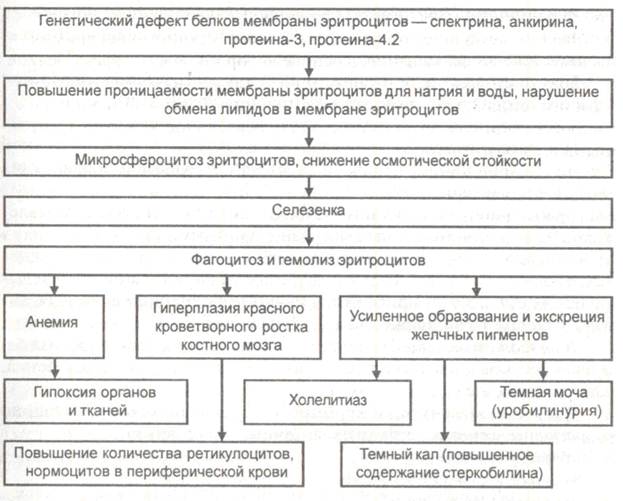
Схема 23-1. Патогенез наследственной микросфероцитарной анемии
(Окороков А.Н., 2001)
Эритроциты обычно становятся гиперхромными. Генетический дефект мембраны эритроцита (потеря мембранных липидов и др.) приводит к повышению ее проницаемости для ионов натрия и избыточному поступлению в клетку воды. Эритроциты набухают, изменяют свою форму, становятся сфероцитами с диаметром около 6 мкм и теряют эластичность. В результате нарушается способность эритроцитов деформироваться в мелких кровеносных сосудах (то есть, уменьшается пластичность эритроцитов). При этом отмечается отрыв частей оболочек микросфероцитов с развитием клеточной фрагментации и последующей их деструкцией, особенно, при прохождении их через эти микрососуды. Резко выраженное укорочение продолжительности жизни микросфероцитов (до 12-14 дней) связано также со снижением энергозапасов в них, так как значительная часть энергии расходуется для удаления избытка воды и натрия из этих клеток. В развитии сфероцитоза важное значение придают селезенке, так как спленэктомия резко уменьшает сфероцитоз.
Наследственные мембранопатии могут также проявляться развитием овалоцитоза, стоматоцитоза, акантоцитоза и т.д.
Наследственные энзимопатии. Обусловлены наследственным дефицитом, главным образом, ферментов пентозофосфатного цикла, аэробного гликолиза и системы глутатиона.
Чаще выявляется энзимопатия, связанная с наследственным дефицитом глюкозо-6-фосфатдегидрогеназы (Г-6-ФДГ), которая, как известно, катализирует начальный этап пентозофосфатного шунта. Заболевание развивается по рецессивному типу, сцепленному с Х-хромосомой, у каждого 10 мужчины, особенно в субтропиках и тропиках. При дефиците Г-6-ФДГ отмечается блокада окисления глюкозо-6-фосфата, главным образом, в пентозофосфатном цикле. Страдает также аэробный гликолиз. Это приводит к нарушению процессов восстановления НАДФ в НАДФН2 и снижению образования восстановленного глутатиона в эритроцитах. Последний защищает SH-группы глобина и мембраны эритроцитов от оксидантного повреждения, в частности, от окислительного действия перекисей и свободных радикалов, образующихся при различной инфекционно-токсической патологии, в том числе при приеме лекарств-окислителей (противомалярийных средств: хинина, делагила; сульфаниламидов, нитрофуранов; производных салициловой кислоты: аспирина; анальгетиков: анальгина, амидопирина; противотуберкулезных средств: ПАСК, фтивазида; и др.). При употреблении в пищу конских бобов и стручковых растений, а также при гриппе и вирусном гепатите может развиваться гемолитический криз. Активизация процессов перекисного окисления гемоглобина и липидов мембран эритроцитов повышает проницаемость последних. Это сопровождается нарушением содержания ионов в эритроцитах, снижением их осмотической резистентности, развитием разрушения гемоглобина и в конечном итоге - острого внутрисосудистого гемолиза. Характерным признаком данного вида анемии является образование в эритроцитах телец Гейнца (денатурированных фрагментов осевшего гемоглобина), которые закрепляясь на внутренней поверхности оболочки в еще большей степени снижают ее эластические свойства. Образование телец Гейнца сопровождается повышением концентрации метгемоглобина в неполноценных эритроцитах.
Выявляют также энзимопатии, связанные с наследственным дефицитом ферментов гликолиза (пируваткиназы, гексокиназы, фосфофруктокиназы и др.) и негликолитических энзимов (глутатион пероксидазы, глутатион синтетазы, глутатион редуктазы).Недостаток данных ферментов нарушает мобилизацию глюкозы и выработку энергии в эритроцитах, что приводит к нарушению ионного состава в этих клетках и снижению их осмотической резистентности и продолжительности жизни.
Наследственные гемоглобинопатии (гемоглобинозы). Это заболевания, в основе которых лежит нарушение синтеза гемоглобина, возникающее в результате наследственно обусловленной аномалии образования глобина, особенно в странах с жарким климатом.
Как известно, у здорового человека HbA1 (около 97 %) содержит две a- и две b-цепи, HbA2 (около 2 %) - две a- и две d - цепи, HbF (около 1 %) - две a- и две g -цепи.
Выделяют качественные и количественные Hb-патии.
Качественные гемоглобинопатии. К ним относят заболевания, связанные с генетическим дефектом структуры (например, аминокислотной последовательности) той или иной цепи глобина (a, b, d, g). Среди них наибольшее клиническое значение представляет гемоглобиноз S или серповидно-клеточная анемия. Данная анемия наследуется с неполным доминированием вследствие мутации структурного гена и синтеза в результате этого не HbA, а HbS, в b-цепи которого у места связывания гена и глобина, гидрофильная глутаминовая кислота заменена гидрофобным валином. В процессе перехода от окисленной к восстановленной форме изменяется заряд патологического HbS, уменьшается его растворимость, происходит образование кристаллических агрегатов, что и лежит в основе серповидности эритроцитов. Серповидные эритроциты (или дрепаноциты) повышают вязкость крови, замедляют кровоток, вызывают стаз, что в еще большей степени углубляет гипоксию. Эти клетки имеют короткий период жизни и быстро разрушаются. Изменение формы эритроцитов и снижение эластичности их мембран определяет повышенное разрушение этих клеток в организме (приемущественно в селезенке, печени)
У гетерозигот по HbS (встречается у 8-13% негров) развивается средней или легкой степени тяжести анемия. Это обусловлено тем, что у них эритроциты содержат 20-45% HbS и 55-80% HbА1. У гетерозигот серповидные эритроциты подвергаются гемолизу, как правило, в ответ на развитие различных видов гипоксии и действие на них различных повреждающих факторов. При гетерозиготной по HbS анемии больные живут относительно долго. В отличие от этого, у гомозигот по HbS (встречается у 0,3% людей) эритроциты имеют только HbS. У таких пациентов серповидные эритроциты в крови разрушаются даже при нормальном рО2 . У них развивается тяжелая гемолитическая анемия, вплоть до развития гемолитических кризов. Такие больные погибают быстрее, они редко доживают до 40 лет.
Количественные гемоглобинопатии. К ним относят наследственные заболевания, в основе которых лежит частичное или полное блокирование, или увеличение синтеза одной из полипептидных (чаще b- или a-) цепей глобина.
При блокадах этих цепей наиболее часто развивается талассемия (средиземноморская анемия).
Блокада синтеза b-цепей глобина проявляется в виде развития b- талассемии. При данной анемии снижается синтез в костном мозге и содержание в крови основного физиологического HbA1, в то время как уровень HbF и HbA2 напротив возрастает. Отмечается уменьшение срока жизни эритроцитов, развиваются хроническая гипохромная анемия и спленомегалия, появляются мишеневидные эритроциты, количество железа в организме обычно в норме или больше нормы. Данная анемия воникает в результате одинаковой или разной степени выраженности мутации одного или двух генов, находящихся в соотвествующих локусах β-глобина на хромосоме 11. В зависимости от степени дефицита β-цепей глобина клинически различают большую β-талассемию (болезнь Кули), промежуточную и малую. Развернутая картина b-талассемии (большая талассемия) возникает при гомозиготной мутации двух генов, что приводит к наибольшему дефициту синтеза β-цепей. При промежуточной β-талассемии выявляется одна слабая, а другая – тяжелая мутация двух генов в соответствующих локусах на 11 хромосоме. При слабой β-талассемии отмечается мутация только одного гена в этой же хромосоме.
Снижение синтезa a-цепей глобина в эритроцитах, возникающее при наследственной патологии соответствующих регуляторных генов обуславливает развитие a- талассемии. Последняя чаще (в 80-85% случаев) возникает в результате мутации одного или нескольких из четырех генов, находящихся в соотвествующих локусах α-глобина на хромосоме 16. клинические проявления коррелируют со степенью нарушения синтеза α-глобиновых цепей. При наибольшей тяжести заболевании угнетается синтез всех нормальных видов гемоглобина, имеющих a-цепь (то есть и HbA1, и HbA2, и HbF).
Избыточное образование b-цепей глобина вызывает у людей образование патологического HbH, который обладает высокой неустойчивостью, легко оседает в клетках, образуя “тельца включения”, и почти полностью не выполняет кислородтранспортной функции. При этом развивается активный гемолиз эритроцитов в макрофагах печени и селезенки. Наследуется данный вид гемоглобинопатии по аутосомно-доминантному типу.
Избыточное образование a-цепей глобина способствует появлению нестабильного гемоглобина, который преципитирует и выпадает в центре эритроцита в виде “телец включения”, придавая им форму мишеней. Кроме того, образующиеся в избытке a-цепи глобина вступают в соединение с SH-груп- пами мембраны эритроцитов и повышают их проницаемость. В результате усиливается гемолиз и развивается неэффективный эритропоэз (значительная часть образовавшихся эритроцитов разрушается уже в костном мозге, в результате чего уменьшается их выход из него.
|
|
Дата добавления: 2015-03-29; Просмотров: 674; Нарушение авторских прав?; Мы поможем в написании вашей работы!