КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Синтез жирных кислот из пальмитиновой кислоты 2 страница
| Типы липопротеинов | Хиломикроны (ХМ) | ЛПОНП | ЛППП | ЛПНП | ЛПВП |
| Состав, % | |||||
| Белки | |||||
| ФЛ | |||||
| ХС | |||||
| ЭХС | |||||
| ТАГ | |||||
| Функции | Транспорт липидов из клеток кишечника(экзогенных липидов) | Транспорт липидов, синтезируемых в печени (эндогенных липидов) | Промежуточная форма превращения ЛПОНП в ЛПНП под действием фермента ЛП-липазы | Транспорт холестерола в ткани | Удаление избытка холестерола из клеток и других липопротеинов. Донор апопротеинов А, С-П |
| Место образования | Эпителий тонкого кишечника | Клетки печени | Кровь | Кровь (из ЛПОНП и ЛППП) | Клетки печени - ЛПВП-пред-шественники |
| Плотность, г/мл | 0,92-0,98 | 0,96-1,00 | 1,00-1,06 | 1,06-1,21 | |
| Диаметр частиц, нМ | Больше 120 | 30-100 | 21-100 | 7-15 | |
| Основные аполипопротеины | В-48 С-П Е | В-100 С-П Е | В-100 Е | В-100 | A-I С-II Е |
В состав ЛПОНП, кроме жиров, входят холестерол, фосфолипиды и белок - апоВ-100. Это очень "длинный" белок, содержащий 11 536 аминокислот. Одна молекула апоВ-100 покрывает поверхность всего липопротеина.
ЛПОНП из печени секретируются в кровь, где на них, как и на ХМ, действует ЛП-липаза. Жирные кислоты поступают в ткани, в частности в адипоциты, и используются для синтеза жиров. В процессе удаления жиров из ЛПОНП под действием ЛП-липазы ЛПОНП сначала превращаются в ЛППП, а затем в ЛПНП. В ЛПНП основными липидными компонентами служат холестерол и его эфиры, поэтому ЛПНП являются липопротеинами, доставляющими холестерол в периферические ткани. Глицерол, освободившийся из липопротеинов, кровью транспортируется в печень, где опять может использоваться для синтеза жиров.
66. Депонирование и мобилизация жиров в жировой ткани. Регуляция синтеза и мобилизации жиров. Роль инсулина, глюкагона и адреналина.
Какой процесс будет преобладать в организме - синтез жиров (липогенез) или их распад (липолиз), зависит от поступления пищи и физической активности. В абсорбтивном состоянии под действием инсулина происходит липогенез, в постабсорбтивном состоянии - липолиз, активируемый глюкагоном. Адреналин, секреция которого увеличивается при физической активности, также стимулирует липолиз.
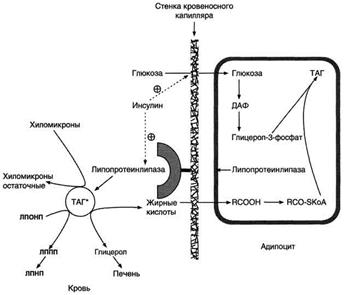
Регуляция синтеза жиров. В абсорбтивный период при увеличении соотношения инсулинглюкагон в печени активируется синтез жиров. В жировой ткани индуцируется синтез ЛП-липазы в адипоцитах и осуществляется её экспонирование на поверхность эндотелия; следовательно, в этот период увеличивается поступление жирных кислот в адипоциты. Одновременно инсулин активирует белки-переносчики глюкозы - ГЛЮТ-4. Поступление глюкозы в адипоциты и гликолиз также активируются. В результате образуются все необходимые компоненты для синтеза жиров: глицерол-3-фосфат и активные формы жирных кислот. В печени инсулин, действуя через различные механизмы, активирует ферменты путём дефосфорилирования и индуцирует их синтез. В результате увеличиваются активность и синтез ферментов, участвующих в превращении части глюкозы, поступающей с пищей, в жиры. Это - регуляторные ферменты гликолиза, пируватдегидрогеназный комплекс и ферменты, участвующие в синтезе жирных кислот из ацетил-КоА. Результат действия инсулина на обмен углеводов и жиров в печени - увеличение синтеза жиров и секреция их в кровь в составе ЛПОНП. ЛПОНП доставляют жиры в капилляры жировой ткани, где действие ЛП-липазы обеспечивает быстрое поступление жирных кислот в адипоциты, где они депонируются в составе триацилглицеринов. Запасание жиров в жировой ткани - основная форма депонирования источников энергии в организме человека. Запасы жиров в организме человека массой 70 кг составляют 10 кг, но у многих людей количество жиров может быть значительно больше. Жиры образуют в адипоцитах жировые вакуоли. Жировые вакуоли иногда заполняют значительную часть цитоплазмы. Скорость синтеза и мобилизации подкожного жира происходит неравномерно в разных частях организма, что связано с неодинаковым распределением рецепторов гормонов на адипоцитах.
Регуляция мобилизации жиров. Мобилизация депонированных жиров стимулируется глюкагоном и адреналином и, в меньшей степени, некоторыми другими гормонами (соматотроп-ным, кортизолом). В постабсорбтивный период и при голодании глюкагон, действуя на адипоциты через аденилатциклазную систему, активирует протеинкиназу А, которая фосфо-рилирует и, таким образом, активирует гормончувствительную липазу, что инициирует липо-лиз и выделение жирных кислот и глицерина в кровь. При физической активности увеличивается секреция адреналина, который действует через β-адренергические рецепторы адипоцитов, активирующие аденилатциклазную систему. В настоящее время обнаружено 3 типа β-рецепторов: β1, β2, β3, активация которых приводит к липолитическому действию. К наибольшему липолитическому действию приводит активация β3-рецепторов. Адреналин одновременно действует и на α2-рецепторы адипоцитов, связанные с ингибирующим G-белком, что инактивирует аденилатциклазную систему. Вероятно, действие адреналина двояко: при низких концентрациях в крови преобладает его антилиполитическое действие через α2-рецепторы, а при высокой - преобладает липолитическое действие через β-рецепторы.Для мышц, сердца, почек, печени при голодании или физической работе жирные кислоты становятся важным источником энергии. Печень перерабатывает часть жирных кислот в кетоновые тела, используемые мозгом, нервной тканью и некоторыми другими тканями как источники энергии. В результате мобилизации жиров концентрация жирных кислот в крови увеличивается приблизительно в 2 раза, однако абсолютная концентрация жирных кислот в крови невелика даже в этот период. Т1/2 жирных кислот в крови тоже очень мал (менее 5 мин), что означает существование быстрого потока жирных кислот из жировой ткани к другим органам. Когда постабсорбтивный период сменяется аборбтивным, инсулин активирует специфическую фосфатазу, которая дефосфорилирует гормончувствительную липазу, и распад жиров останавливается.

| Кровоток |
67.Основные фосфолипиды и гликолипиды тканей человека (глицерофосфолипиды, сфингофосфолипиды, гликоглицеролипиды, гликосфиголипиды). Представление о биосинтезе и катаболизме этих соединений.
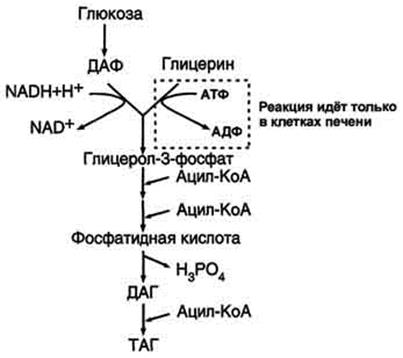
Синтез фосфатидилхолинов, фосфатидилэтаноламинов и фосфатидилсеринов. Начальные этапы синтеза глицерофосфолипидов и жиров происходят одинаково до образования фосфатидной кислоты. Фосфатидная кислота может синтезироваться двумя разными путями: через глицеральдегид-3-фосфат и через дигидроксиацетонфосфат

На следующем этапе фосфатидаза отщепляет от фосфатидной кислоты фосфатный остаток, в результате чего образуется диацилглицерол. Дальнейшие превращения диацилглицерола также могут идти разными путями. Один из вариантов - образование активной формы "полярной головки" фосфолипида: холин, серии или этаноламин превращаются в ЦДФ-холин, ЦДФ-серин или ЦДФ-этаноламин. Далее диацилглицерол взаимодействует с ЦМФ-производными, при этом выделяется ЦМФ, и образуется соответствующий фосфолигид, например фосфатидилхолин. Между глицерофосфолипидами возможны различные взаимопревращения. Фосфатидилхолин может образовываться и другим путём: из фосфатидилэтаноламина, получая последовательно 3 метальные группы от SAM. Фосфатидилсерин может превращаться в фосфатидилэтаноламин путём декарбоксилирования. Фосфатидилэтаноламин может превращаться в фосфатидилсерин путём обмена этаноламина на серии.
Сфинголипиды - производные церамида, образующегося в результате соединения аминоспирта сфингозина и жирной кислоты. В группу сфинголипидов входят сфингомиелины и гликосфинголипиды фингомиелины находятся в мембранах клеток различных тканей, но наибольшее их количество содержится в нервной ткани. Сфингомиелины миелиновых оболочек содержат в основном жирные кислоты с длинной цепью: лигноцери-новую (24:0) и нервоновую (24:1) кислоты, а сфингомиелин серого вещества мозга содержит преимущественно стеариновую кислоту.
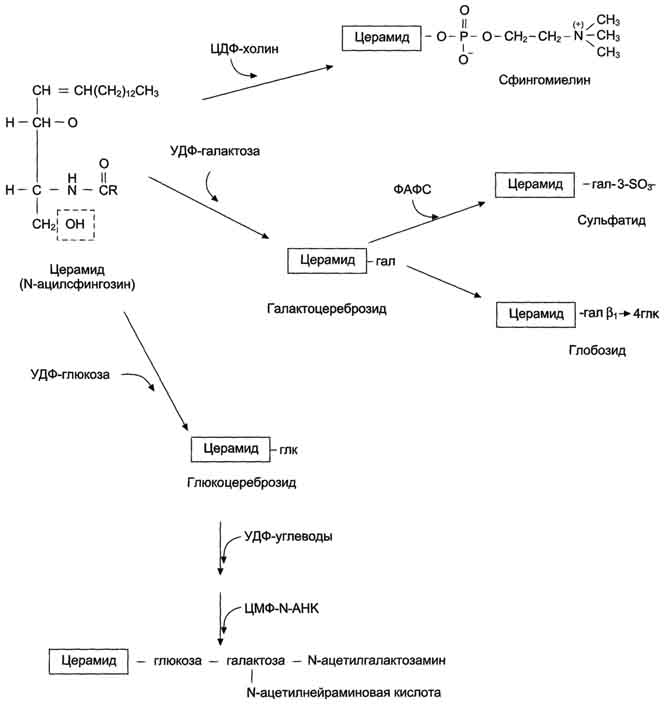
Гликосфинголипиды - гликолипиды, в состав которых входят церамид и один или несколько остатков углеводов, и сиаловая (N-ацетилнейраминовая) кислота узнавание клеток и их взаимодействие. Интересно, что углеводная часть структуры антигенов на поверхности эритроцитов (по системе АВО) может быть связана как с церамидом, так и с белками. В последнем случае структура антигена является не гликолипидом, а гликопротеином.
Некоторые ганглиозиды - рецепторы бактериальных токсинов. Например, GMl, находящийся на поверхности клеток кишечного эпителия, является местом прикрепления холерного токсина - белка, секретируемого возбудителями холеры.
Функции гликосфинголипидов можно суммировать следующим образом:
Взаимодействие между:
- клетками;
- клетками и межклеточным матриксом;
- клетками и микробами.
Модуляция:
- активности протеинкиназ;
- активности рецептора фактора роста;
- антипролиферативного действия (апоптоза, клеточного цикла).
Обеспечение:
- структурной жёсткости мембран;
- конформации белков мембран.
Синтез церамида и его производных. Синтез сфинголипидов начинается с образования церамида. Серии конденсируется с пальмитоил-КоА. Продукт их взаимодействия сначала восстанавливается коферментом NADPH, затем к аминогруппе дигидросфингозина амидной связью присоединяется жирная кислота, содержащая, как правило, 24 атома углерода. После окисления FAD-зависимой дегидрогеназой образуется церамид. Церамид служит предшественником в синтезе большой группы сфинголипидов: сфингомиелинов, не содержащих углеводов, и гликосфинголипидов. Последующие реакции синтеза катализируются специфическими трансферазами, набор которых отличается в разных тканях. Соединение фосфорилхолина с церамидом сфингомиелин-синтазой приводит к образованию сфингомие-лина. Присоединение углеводных компонентов катализируется специфическими гликозилтрансферазами. Донорами углеводных компонентов служат активированные сахара: УДФ-галактоза и УДФ-глюкоза. Галактоцереброзид - главный липид миелиновых оболочек; глюкоцереброзид входит в состав мембран многих клеток и служит предшественником в синтезе более сложных гликолипидов или продуктом на пути их катаболизма.
Катаболизм сфингомиелина. В лизосомах находятся ферменты, способные гидролизовать любые компоненты клеток. Эти ферменты называют кислыми гидролазами, так как они активны в кислой среде. Значение рН = 5, оптимальное для работы ферментов, создаётся протонным насосом, который, используя энергию АТФ, накачивает ионы водорода в лизосомы. Катаболизм сфингомиелинов и гликолипидов происходит в лизосомах. В распаде сфингомиелинов участвуют 2 фермента - сфингомиелиназа, отщепляющая фосфорилхолин, и церамидаза, продуктами действия которой являются сфингозин и жирная кислота
Катаболизм гликосфинголипидов. Катаболизм гликосфинголипидов начинается с перемещения их с поверхности клетки в цитоплазму по механизму эндоцитоза. В результате молекулы, расположенные на поверхности мембран, оказываются в эндоцитозных везикулах в цитоплазме и сливаются с лизосомами. В лизосомах находятся все ферменты, необходимые для гидролиза сложных молекул гликосфинголипидов: α- и β-галактозидазы, β-глюкозидазы, нейраминидаза (сиалидаза) и церамидаза. В результате последовательных реакций гидролиза сложные молекулы гликосфинголипидов распадаются до мономеров: глюкозы, галактозы, жирной кислоты, сфингозина и других метаболитов.
68.Нарушение обмена нейтрального жира (ожирение), фосфолипидов и гликолипидов. Сфинголипидозы
Ожирением считают состояние, когда масса тела превышает 20% от "идеальной" для данного индивидуума. Образование адипоцитов происходит ещё во внутриутробном состоянии, начиная с последнего триместра беременности, и заканчивается в препубертатный период. После этого жировые клетки могут увеличиваться в размерах при ожирении или уменьшаться при похудании, но их количество не изменяется в течение жизни.
Первичное ожирение. Первичное ожирение характеризуется множеством гормональных и метаболических особенностей у лиц, страдающих этим заболеванием. В самом общем виде можно сказать, что первичное ожирение развивается в результате алиментарного дисбаланса - избыточной калорийности питания по сравнению с расходами энергии. Суточные потребности организма в энергии складываются из:
- основного обмена - энергии, необходимой для поддержания жизни; основной обмен измеряют по поглощению кислорода или выделению тепла человеком в состоянии покоя утром, после 12-часового перерыва в еде;
- энергии, необходимой для физической активности.
Затраты энергии, необходимые для физической активности, разделяют на 3 уровня:
· I - 30% энергии от основного обмена (у людей, ведущих сидячий образ жизни);
· II - 60-70% от энергии основного обмена (у людей, которые 2 ч в день имеют умеренную физическую нагрузку);
· III - 100% и более от энергии основного обмена (у людей, которые в течение нескольких часов в день занимаются тяжёлой физической работой).
В зависимости от интенсивности нагрузки и возраста суточная потребность в энергии колеблется у женщин от 2000 до 3000 ккал в день, а у мужчин - от 2300 до 4000 ккал. Количество потребляемой пищи определяется многими факторами, в том числе и химическими регуляторами чувства голода и насыщения. Эти чувства определяются концентрацией в крови глюкозы и гормонов, которые инициируют чувство насыщения: холецистокинина, нейротензина, бомбезина, лептина.
Причины первичного ожирения:
- генетические нарушенвся (до 80% случаев ожирения - результат генетических нарушений);
- состав и количество потребляемой пищи, метод питания в семье;
- уровень физической активности;
- психологические факторы.
Генетические факторы в развитии ожирения. Метаболические различия между тучными и худыми людьми до настоящего времени не могут быть определены однозначно. Существует несколько теорий, объясняющих эти различия:
· генетически детерминированная разница в функционировании "бесполезных" циклов (субстратных циклов, раздел 7). Эти циклы состоят из пары метаболитов, превращаемых друг в друга с помощью двух ферментов. Одна из этих реакций идёт с затратой АТФ. Например:
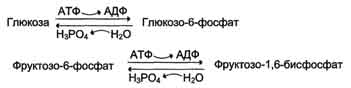
· если эти субстраты превращаются друг в друга с одинаковой скоростью, то происходит "бесполезный" расход АТФ и, соответственно, источников энергии, например жиров;
· у людей, склонных к ожирению, вероятно, имеется более прочное сопряжение дыхания и окислительного фосфорилирования, т.е. более эффективный метаболизм;
· возможно, разное соотношение аэробного и анаэробного гликолиза. Анаэробный гликолиз (как менее эффективный) "сжигает" гораздо больше глюкозы, в результате снижается её переработка в жиры;
· у отдельных ивдивидуумов имеется различие в активности Nа+/К+-АТФ:азы, работа которой требует до 30% энергии, потребляемой клетками.
У человека и животных имеется " ген ожирения " - obese gene (ob). Продуктом экспрессии этого гена служит белок лептин, состоящий из 167 аминокислот, который синтезируется и секретируется адипоцитами и взаимодействует с рецепторами гипоталамуса. В результате его действия снижается секреция нейропептида Y. Нейропептид Y стимулирует пищевое поведение, поиск и потребление пищи у животных. Другие пептиды, участвующие в регуляции чувства сытости, например холецистокинин, также влияют на секрецию нейропептида Y. Таким опосредованным путём лептин выступает регулятором жировой массы, необходимой для роста и репродукции. Уровень лептина у больных ожирением может быть различным. У 80% больных концентрация лептина в крови тучных людей больше в 4 раза, чем у людей с нормальной массой тела. В этих случаях имеется генетический дефект рецепторов лептина в гипоталамусе, поэтому, несмотря на продукцию лептина, центр голода в гипоталамусе продолжает секрецию нейропептида Y. 20% больных имеют изменения в первичной структуре лептина. К настоящему времени описаны 5 одиночных мутаций в гене лептина, которые приводят к развитию ожирения. У этих больных наблюдают повышение отложения жиров в жировой ткани, чрезмерное потребление пищи, низкую физическую активность и развитие сахарного диабета типа II. Патогенез ожирения при дефекте гена ob может быть следующим: низкий уровень лептина в крови служит сигналом недостаточного количества запаса жиров в организме; этот сигнал включает механизмы, приводящие к увеличению аппетита и в результате к увеличению массы тела. Следовательно, можно сделать вывод о том, что первичное ожирение - не просто следствие переедания, а результат действия многих факторов, т.е. ожирение - полигенное заболевание.
 Вторичное ожирение - ожирение, развивающееся в результате какого-либо основного заболевания, чаще всего эндокринного. Например, к развитию ожирения приводят гипотиреоз, синдром Иценко-Кушинга, гипогонадизм и многие другие заболевания
Вторичное ожирение - ожирение, развивающееся в результате какого-либо основного заболевания, чаще всего эндокринного. Например, к развитию ожирения приводят гипотиреоз, синдром Иценко-Кушинга, гипогонадизм и многие другие заболевания
Генетический дефект сфингомиелиназы - причина болезни Ниманна-Пика. Дети с таким дефектом погибают в раннем возрасте. Симптомы заболевания: увеличение печени и селезёнки (гепатоспленомегалия), в лизосомах которых накапливается сфингомиелин; умственная отсталость. Генетический дефект другого фермента (церамидазы) приводит к развитию болезни Фарбера, симптомами которой также являются гепато- и спленомегалия, а также поражение суставов (болезненность, отёчность).
Генетические дефекты лизосомных ферментов катаболизма гликосфинголипидов. В норме синтез и катаболизм гликосфинголипидов сбалансированы таким образом, что количество этих компонентов в мембранах постоянно. Если имеется генетический дефект какого-либо лизосомного фермента, участвующего в катаболизме гликосфинголипида, то в лизосомах накапливается не-деполимеризованный субстрат, так называемые "остаточные тельца", размеры лизосом увеличиваются, их мембрана может разрушаться, ферменты выходят в цитозоль, и функции клеток нарушаются. Генетические заболевания вследствие дефекта какого-либо из ферментов катаболизма гликосфинголипидов называют сфинголипидоза-ми, или лизосомными болезнями. Эти заболевания редки, но среди некоторых популяций людей их частота очень высока. Так, болезнь Гоше вследствие дефекта фермента β-глюкрзидазы у евреев встречается с частотой 166:100 000, болезнь Тея-Сакса (дефект фермента β-гексозаминидазы) - с частотой 33:100 000. Сфинголипидозы обычно приводят к смерти в раннем возрасте, так как происходит поражение клеток нервной ткани, где сконцентрированы гликосфинголипиды. Однако при болезнях Гоше и Фабри больные живут, относительно долго.
Сфинголипиды, метаболизм: заболевания сфинголипидозы, таблица
| Заболевание | Фермент, недостаточностькоторого обусловливает заболевание | Накапливающийся:липид: | Клинические симптомы |
| Фукозидоз | альфа-Фукозидаза | Cer-Glc-GalNAc-Cal-:-Fuc Н-Изоантиген | Слабоумие, спастическое состояние мышц, утолщение кожи |
| Генерализованный ганглиозидоз | GM1-бета-Галактозидаза | Cer-Glc-Gal(NeuAc)-GalNAc-:-Gal Ганглиозид GM1 | Умственная отсталость, увеличениепечени, деформация скелета |
| Болезнь Тея-Сакса | Гексозаминидаза А | Cer-Glc-Gal-(NeuAc)-:-GalNAc Ганглиозид GM2 | Умственная отсталость, слепота, мышечная слабость |
| Вариант болезни Тея-Сакса, или болезнь Сандхоффа | Гексозаминидазы А и В | Cer-Glc-Gal-Gal-:-GalNAc Глобозид + ганглиозид GM2 | Те же, что и в случае болезни Тея-Сакса, но развиваются быстрее |
| Болезнь Фабри (сильно выражена только у мужчин); рецессивный генетический признак, связан с X-хромосомой | альфа-Галактозидаза | Cer-Glc-Gal-:-Gal Глоботриаозилцерамид | Кожная сыпь, почечная недостаточность |
| Церамидлактозидлипидоз | Церамидлактозидаза (бета-галактозидаза) | Cer-Glc-:-Gal Церамидлактозид | Прогрессирующее поражение мозга,увеличение печени и селезенки |
| Метахроматическая лейкодистрофия | Арилсульфатаза | Cer-Gal-:-OSO3 3-Сульфогалактозилцерамид | Умственная отсталость и психические нарушения у взрослых;демиелинизация |
| Болезнь Краббе | бета-Галактозидаза | Cer-:-Gal Галактозилцерамид | Умственная отсталость, почти полное отсутствие миелина |
| Болезнь Гоше | бета-Глюкозидаза | Cer-:-Glc Глюкозилцерамид | Увеличение печени и селезенки, эрозия трубчатых костей, умственная отсталость у детей |
| Болезнь Нимана-Пика | Сфингомиелиназа | Cer-:-P-холин Сфингомиелин | Увеличение печени и селезенки, умственная отсталость; фатальна в раннем возрасте |
| Болезнь Фарбера | Церамидаза | Ацил-:-Сфингозин Церамид | Хрипота, дерматит, деформация скелета, умственная отсталость; фатальна в раннем возрасте |
Обозначения: NeuAc - N-ацетилнейраминовая кислота; Cer - церамид; Glc - глюкоза; Gal - галактоза; Fuc - фукоза
69.Строение и биологические функции эйкозаноидов. Биосинтез простагландинов и лейкотриенов.
Эйкозаноиды, включающие в себя простагландины, тромбоксаны, лейкотриены и ряд других веществ, - высокоактивные регуляторы клеточных функций. Они имеют очень короткий Т1/2, поэтому оказывают эффекты как "гормоны местного действия", влияя на метаболизм продуцирующей их клетки по аугокзэинному механизму, и на окружающие клетки - по паракринному механизму. Эйкозаноиды участвуют во многих процессах: регулируют тонус ГМК и вследствие этого влияют на АД, состояние бронхов, кишечника, матки. Эйкозаноиды регулируют секрецию воды и натрия почками, влияют на образование тромбов. Разные типы эйкозаноидов участвуют в развитии воспалительного процесса, происходящего после повреждения тканей или инфекции. Такие признаки воспаления, как боль, отёк, лихорадка, в значительной мере обусловлены действием эйкозаноидов. Избыточная секреция эйкозаноидов приводит к ряду заболеваний, например бронхиальной астме и аллергическим реакциям.
Структура, номенклатура и биосинтез простагландинов и тромбоксанов Хотя субстраты для синтеза эйкозаноидов имеют довольно простую структуру (полистовые жирные кислоты), из них образуется большая и разнообразная группа веществ. Наиболее распространены в организме человека простагландины, которые впервые были выделены из предстательной железы, откуда и получили свое название. Позже было показано, что и другие ткани организма синтезируют простагландины и другие эйкозаноиды.
|
|
Дата добавления: 2015-04-24; Просмотров: 1605; Нарушение авторских прав?; Мы поможем в написании вашей работы!