КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Структура комплексних сполук
Вступ
1. 1.Еквівалент. Закон еквівалентів
Закон еквівалентів — маси речовин, які вступають у реакцію та утворюються після неї, пропорційні їхнім еквівалентам.
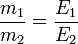
Із закону сталості складу випливає, що речовини взаємодіють між собою і утворюють продукти у певних, чітко визначених (еквівалентних) кількостях.
Еквівалент (Е) — еквівалентом елемента називають таку його кількість, яка взаємодіє з 1 м.ч. Н або 8 м.ч. О або заміщує таку кількість хімічних реакцій.
Еквівалент - це частина атому, молекули або йону яка припадає на одну зміної валентності або ступеню окиснення. Наприклад, в сполуках HCl, H2S, NH3, CH4 еквівалент хлора, сірки, нітрогену, вуглеця дорівнює відповідно 1 моль, 1/2 моль, 1/3 моль, 1/4 моль.
Еквівалент речовини – це реальна або умовна частка цієї речовини, яка в кислотно-основній реакції звільняє один гідроген (1+)- іон або сполучається з ним, а в окисно-відновній реакції приєднує або звільняє один електрон.
Еквівалент речовини змінний і залежить від реакції, у якій ця речовина бере участь.
Маса одного еквіваленту називається його еквівалентною масою. Так, в наведених вище прикладах еквівалентні маси хлору, сірки, нітрогену та вуглецю відповідно дорівнюють 34,45 г/моль, 32/2=16 г/моль, 14/3=4,67 г/моль, 12/4=3 г/моль.
Число, яку показує, яка частка реальної частинки речовини X еквівалентна одному гідроген (1+)- іону в даній кислотно-основній реакції, називається фактором еквівалентності речовини X. Фактор еквівалентності — це величина безрозмірна.
2.Використання стахіометричних законів для розрахунків.
Стехіометрія — розділ хімії про співвідношення реагентів в хімічних реакціях.
Дозволяє теоретично обчислювати необхідні маси та об'єми реагентів.
В основі стехіометрії лежать закони збереження маси, еквівалентів, Авогадро, Гей-Люссака, сталості складу, кратних відносин.
Всі закони стехіометрії обумовлені атомно-молекулярною будовою речовини.
Термін " стехіометрія " ввів І. Ріхтер в 1793 році.
Відносини кількостей реагентів, рівні відносинам коефіцієнтів у стехіометричному рівнянні реакції, називаються стехіометричними. Якщо речовини реагують у співвідношенні 1:1, то їхні відповідні кількості називають еквімолярними.
Речовини, для яких спостерігаються відхилення від законів стехіометрії, звуться нестехіометричними. Відхилення від законів стехіометрії спостерігаються для конденсованих фаз і пов'язані з утворенням твердих розчинів (для кристалічних речовин), з розчиненням в рідині надлишку компонента реакції або термічною дисоціацією з'єднання, що утворюється (в рідкій фазі, в розплаві). Закони стехіометрії використовують у розрахунках, пов'язаних з формулами речовин і знаходженням теоретично можливого виходу продуктів реакції.
Самостійна робота №2
Періодичний закон і періодична система хімічних елементів
1.Закономірності зміни властивостей елементів у періодичній системі.
Хімічні властивості елементів (а вже тим більше їх сполук!) Безпосередньо залежать від будови атома.
Не треба вчити напам'ять хімічні властивості кожного атома, не треба зазубрювати хімічні реакції... відповідь на будь-яке питання з хімії знаходиться в періодичною системою елементів.
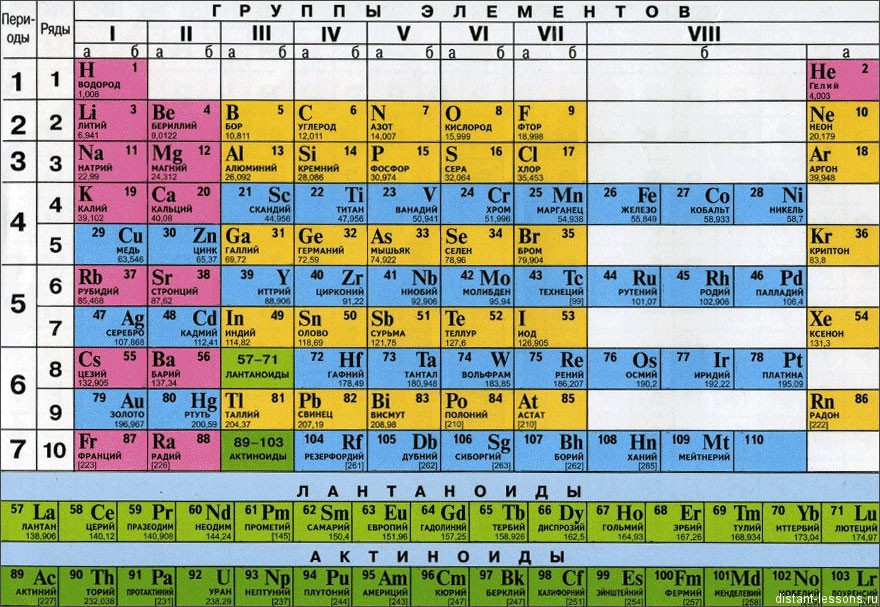

Зміни властивостей хімічних елементів та їх сполук в групах:
У групах всі елементи мають подібне електронне будова.
Відмінностей в наповненні зовнішнього енергетичного рівня електронами немає.
Змінюється розмір атома - зверху вниз в групі радіуси атомів збільшуються!
1) зовнішні електрони все слабше притягуються до ядра атома;
2) зростає здатність атома віддавати електрони.
3) здатність віддавати електрони = металеві властивості, тобто
закономірність зміни хімічних властивостей елементів і їх сполук в групах:
У групах зверху вниз зростають металеві властивості елементів
посилюються основні властивості їх сполук
Зміни хімічних властивостей елементів і їх сполук в періодах:
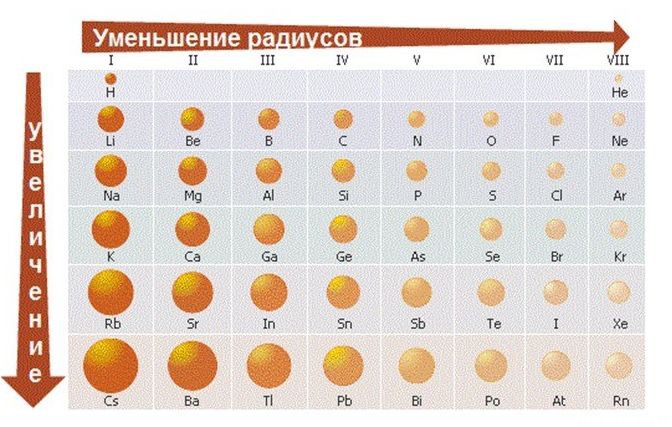
В періодах спостерігається дещо інша картина:
1) Зліва направо в періодах радіуси атомів зменшуються;
2) кількість електронів на зовнішньому шарі при цьому збільшується;
3) електронегативність елементів = неметалеві властивості збільшується
закономірності зміни хімічних властивостей елементів і їх сполук в періодах:
В періодах зліва направо зростають неметалеві властивості елементів, електронегативність;
посилюються кислотні властивості їх сполук

Виходячи з цих міркувань виходить, що звання «Король неметалла» у нас присуджується... (барабанний дріб)... F! Поруч з ним навіть кисень (O) проявляє позитивну ступінь окислення: OF2 - безбарвний отруйний газ з неприємним запахом.
Отже, підведемо підсумок:
Із збільшенням заряду ядра атомів спостерігається поступове закономірне зміна властивостей елементів і їх сполук від металевих до типово неметаллическим, що пов'язано зі збільшенням числа електронів на зовнішньому енергетичному рівні.
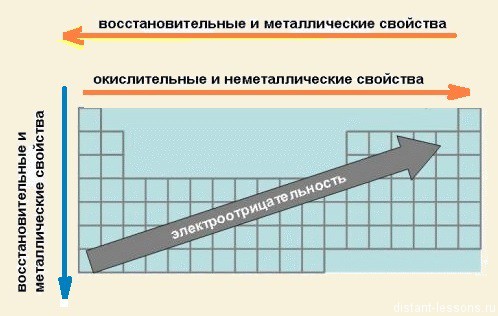
Є ще елементи, які утворюють так звані амфотарні з'єднання. Вони проявляють як металеві, так і неметалеві властивості.
2.Загальнонаукове та філософське значеня періодичного закону.
Філософське значення періодичного закону Д. І. Менделєєва
Відкриття періодичного закону було підготовлено всім попереднім розвитком хімії, головним чином атомно-молекулярної теорією і вченням про хімічних елементах. Науковими передумовами послужили встановлення близьких до сучасних атомних мас багатьох хімічних елементів, і вчення про атомности (валентності), і про хімічній сполуці (засноване на унітарних і молекулярних уявленнях). «Зв'язавши поняття про хімічні елементи новими узами з Дальтоновим вченням про кратному або атомному складі тел, періодичний закон відкрив в природної філософії нову область для мислення. Канту здавалося, що в світі є «два предмета, постійно викликають людське здивування і благоговіння: моральний закон всередині нас і зоряне небо над нами». Вдумуючись в природу елементів і в періодичний закон, слід сюди приєднати третій предмет: «природу елементарних індивідуумів - поруч фактів усюди виражену», так як без них немислимо саме зоряне небо і так як в атомах одноразово відкривається і своєрідність індивідуальностей, і безмежна повторюваність особин, і підпорядкованість удаваного свавілля індивідуумів загальному гармонійному порядку природи».
Таким чином, періодичний закон був підсумковим ланкою в ланцюзі наукових відкриттів, яка деяким історикам здається єдино логічною. Центральними в логічній схемі є поняття про елементи-аналогах і вчення про форму сполук. Аналогія в хімії та індивідуальність хімічних елементів виявилися сполученими з поняттям про елементи-аналогах, яке оформилося в першій половині XIX в.. При цьому хімічна аналогія розглядалася як результат внутрішнього подібності атомів.
Як метод пізнання аналогія використовувалася й раніше. Багато понять в хімії були тісно пов'язаними з нею (гемолітичні ряди органічних сполук, ізомерія, ізоморфізм). Все це були різні випадки аналогії, хоча ознаки подібності та відмінності і причини їх появи були неоднаковими, що відбивалося па класифікації об'єктів. Методологічний підхід Д. І. Менделєєва при вживанні поняття «еходетво» показує, що, по-перше, воно багатозначно, по-друге, сама можливість виділення ознак для побудови судженні, але аналогії свідчить про певний рівень стану хімічної науки. Відомо, що аналогія виправдовується, а метод судження по аналогії виявляється плідним тоді, коли істотні ознаки предметів або явищі, поставлених в ряд порівняння, збігаються. Але саме внутрішні ознаки нерідко приховані від дослідника, що дуже часто зустрічається в хімії.
Філософське значення вчення про періодичність це перехід від стадії специфічного, властивого тій чи іншій сукупності елементів, до стадії загального, характерного для всіх елементів: «... визнають надто багато індивідуальним... пов'язати ці індивідуальності загальної идеею мета моєї природної системи».
Подальше поширення принципу періодичності, сформульованого видатним російським ученим, показало як величезні досягнення в його застосуванні, так і певні неузгодження.
Вивчення характеру зміни численних властивостей приводили до висновку: «... періодична змінюваність простих і складних тіл підкоряється деякому вищому закону, природу якого, і тим більше причину нині ще немає коштів охопити. Цілком ймовірно, вона криється в основних засадах «внутрішньої механіки атомів і частинок»
Періодичний закон має величезне природничонаукове і філософське значення. Він дозволив розглядати всі елементи в їх взаємному зв'язку і прогнозувати властивості невідомих елементів. Завдяки Періодичному закону багато наукових пошуки (наприклад, в галузі вивчення будови речовини - в хімії, фізиці, геохімії, космохімії, астрофізиці) отримали цілеспрямований характер. Періодичний закон - яскравий прояв дії загальних законів діалектики, зокрема закону переходу кількості в якість.
Періодичний закон Менделєєва, фундаментальний закон, який встановлює періодичну зміну властивостей хімічних елементів в залежності від збільшення зарядів ядер їх атомів. Відкрито Д. І. Менделєєвим в 1869 при зіставленні властивостей всіх відомих у той час елементів і величин їх атомних ваг.
У природі (на Землі) елементи важче урану, що має порядковий номер 92 в таблиці, не зустрічаються, так як є радіоактивними і їх ядра вже розпалися за більш ніж чотири мільярди років Земний історії. Всі елементи важче урану синтезуються в спеціальних ядерних реакторах, в тому числі і плутоній, використаний в роботі американських фізиків.
Наукове значення періодичного закону Д.І. Менделєєва
менделеєв періодична таблиця хімія
Дмитро Іванович Менделєєв народився в лютому 1834 р. у місті Тобольську, у родині директора місцевої гімназії. З 1850 р. навчався на фізико-математичному факультеті Петербурзького педагогічного інституту. У 1855 р. закінчив його з золотою медаллю і був направлений учителем гімназії спочатку в Сімферополь, а потім в Одесу. У 1856 р. Дмитро Менделєєв відправився у Петербург і захистив магістерську дисертацію за темою "Про питомі об'єми", після чого на початку 1857 р. був прийнятий приват-доцентом з кафедри хімії в Петербурзький університет. 1859 - 1861 р. він перебував у науковому відрядженні у Німеччині, у Гейдельберзькому університеті. У 1860 р. Менделєєв взяв участь у роботі першого міжнародного хімічного конгресу в Карлсрує.
У 1861 р. Менделєєв написав перший у Росії підручник з органічної хімії. Навесні 1862 р. підручник був визнаний гідним повної Демидівської премії. У 1863 р. він отримав місце професора у Петербурзькому технологічному інституті, а в 1866 р. - у Петербурзькому університеті, де читав лекції з органічної, неорганічної і технічної хімії. У 1865 р. Менделєєв захистив докторську дисертацію за темою "Про сполуки спирту з водою".
У 1867 р. Менделєєв перейшов у Петербурзький університет на посаду професора хімії і повинен був читати лекції з неорганічної хімії. Однак, на його думку, ні в Росії, ні за рубежем не було курсу загальної хімії, який можна було б рекомендувати студентам. Дмитро Іванович вирішив написати його сам.
Ця робота одержала назву "Основи хімії", і виходила протягом декількох років окремими випусками. Працюючи над другим випуском, Менделєєв зштовхнувся зі складнощами, пов'язаними з послідовністю викладу матеріалу. Спочатку він хотів з групувати всі описувані ним елементи за валентностями, але потім обрав інший метод і об'єднав їх в окремі групи, виходячи з подібності властивостей і атомної ваги. На той час вже були спроби скласти таблиці елементів. Німецький хімік Гмелін, опублікував свою таблицю в 1843 р. У 1857 р. англійський хімік Одлінг запропонував свою. Однак зв'язок груп елементів між собою залишався незрозумілим. Менделєєву вдалося знайти його, розташувавши всі елементи в порядку зростання їхньої атомної маси.
Написавши на окремих картках назви елементів з позначенням їхньої атомної ваги і корінних властивостей, Менделєєв став розкладати їх у різноманітних комбінаціях, переставляючи і змінюючи місцями. Справа ускладнювалася тим, що багато елементів тоді ще не були відкриті, а атомна вага уже відомих визначена з великими похибками. Однак Дмитро Іванович незабаром виявив закономірність. У лютому 1869 р. Менделєєв розіслав російським і закордонним хімікам надрукований на окремому аркуші "Досвід системи елементів, заснований на їхній атомній вазі і хімічній подібності".
Перший варіант періодичної таблиці досить сильно відрізнявся від звичної нам зі школи таблиці Менделєєва. Кілька елементів, як потім виявилося, були в цьому першому варіантові розміщені не за своїми місцями. Однак, зіставляючи властивості елементів, що потрапили у вертикальні стовпчики, можна було ясно бачити, що вони періодично змінюються мірою зростання атомної ваги. Це було найголовніше відкриття Менделєєва. Незбіжність у своєму періодичному ряді Менделєєв пояснив тим, що науці відомі ще не всі хімічні елементи.
Він залишив у таблиці чотири незаповнені клітинки, але спрогнозував їхню атомну вагу і хімічну подібність. Він також виправив неточно визначені атомні маси елементів. Перший начерк таблиці Дмитро Іванович, згодом, піддав корегуванню. Поряд з головними груповими елементами Менделєєв став виділяти підгрупи. Він виправив атомну вагу одинадцяти елементам і змінив місце розташування двадцятьох. У 1871 р. періодична таблиця прийняла цілком сучасний вигляд. Однак, ніхто з відомих європейських хіміків не оцінив важливості зробленого Менделєєвим відкриття.
Ставлення до періодичного закону змінилося тільки в 1875 р., коли був відкритий елемент галій, властивості якого збігалися з прогнозами Менделєєва. Новим тріумфом Менделєєва стало відкриття в 1879 р. скандію, а в 1886 р. германію, властивості яких також відповідали описам Менделєєва.
У наступні роки з-під пера Менделєєва вийшло ще кілька основних праць з різних розділів хімії. Його повна наукова і літературна спадщина величезна і містить 431 роботу. Праці Менделєєва отримали широке міжнародне визнання. Він був обраний членом багатьох академій наук, іноземних наукових товариств. Тільки Російська Академія наук на виборах 1880 р. забалотувала його через внутрішні інтриги.
Самостійна робота 3
Хімічний зв'язок
1.Утворення двохатомних гомоядерних молекул елементів другого періоду за методом ММО
2.Водневий зв'язок. Природа й енергія водневого зв’язку.
ВОДНЕВИЙ ЗВ'ЯЗОК
ПРИРОДА Й ЕНЕРГІЯ ВОДНЕВОГО ЗВ'ЯЗКУ
Водневим зв'язком називають зв'язок двох дуже електронегативних атомів за допомогою атома водню. Перше уявлення про його існування висловив Ільїнський (1887) і теоретично обгрунтували Латімер і Родебуш (1920). Водневий зв'язок (Н-зв'язок) займає проміжне положення між електростатичним притяганням і донорно-акцепторною взаємодією. Він звичайно виникає під час взаємодії сильно полярних молекул між собою або з іншими полярними молекулами:
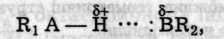
де А та В — атоми електронегативних елементів (крапками позначено водневий зв'язок).
Водневий зв'язок тим міцніший, чим більша електронегатив-ність атома-партнера і чим менший його розмір. Тому він характерний для водневмісних сполук Флюору та Оксигену, меншою мірою — Нітрогену, ще меншою — Хлору, Сульфуру та Карбону.
Утворення водневого зв'язку зумовлене дуже малим розміром додатньо поляризованого атома Гідрогену, який фактично перетворюється на протон, і його здатністю глибоко проникати в електронну оболонку сусіднього від'ємно поляризованого атома. Між протоном та негативно зарядженим атомом полярно-ковалентної молекули виникає електростатична взаємодія. Проте чисто електростатичною взаємодією неможливо пояснити напрямленість і насичуваність водневого зв'язку, залежність між його енергією та дипольним моментом або поляризовністю взаємодіючих атомів. Необхідно ще враховувати й донорно-акцепторну взаємодію, на що вперше вказав Соколов (1964).
В електронній структурі фрагмента А—Н... В з неподіленою електронною парою атома В і двома електронами ковалентного зв'язку А—Н відбувається зміщення валентної електронної густини в напрямку від В до А. Це, природно, стосується не лише атомів А, Н і В, але майже всіх атомів обох взаємодіючих молекул. У результаті між цими молекулами утворюється донорно-акцептор-ний зв'язок. Особливістю цього зв'язку є те, що донор електронів — молекула R2B — має атом В з електронною парою, що безпосередньо взаємодіє з атомом Н групи А—Н. Через цю групу переноситься заряд на акцептор — молекулу R1AH. Чим більше молекул бере участь в утворенні комплексу з водневими зв'язками, тим ефективніше перерозподіляється електронний заряд по ряду послідовно зв'язаних молекул, тим сильніші водневі зв'язки й вища їхня енергія.
Описати електронну конфігурацію водневого зв'язку можна за допомогою схеми рівнів трьохорбітального трицентрового фрагмента А—Н... В з чотирма електронами (два від групи А—Н і два від неподіленої пари атома В). Група А—Н, як двохатомна молекула, має σ i σ* -орбіталі В — орбіталь неподіленої пари. Хвильова функція електрона в Н-зв'язку утворюється як лінійна комбінація цих трьох орбіталей

У комплексах з Н-зв'язком рівні МО, локалізовані в основному на донорі електронів, завжди знижуються, а рівні МО, локалізовані на акцепторі — підвищуються порівняно з рівнями вільних молекул. Це відрізняє Н-зв'язок від ковалентного, де знижуються рівні МО обох учасників.
Водневий зв'язок може виникати не тільки між атомами різних молекул, а й між атомами однієї молекули.
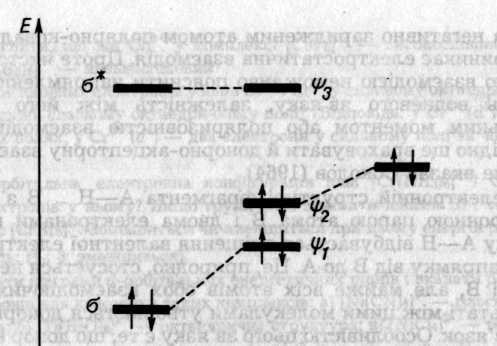
Рис. 6.44. Молекулярні орбіталі акцептора А—Н (зліва) неподіленої пари атома В донора (справа) і водневого містка А—Н —В (у центрі)

Енергія водневого зв'язку в загальному містить у собі різні енергетичні зміни в усій системі й залежить від природи атомів А та В і від електронної конфігурації молекул R1AH і R2B у цілому. Розрізняють слабкі несиметричні та міцні симетричні водневі зв'язки. У слабких зв'язках між'ядерна відстань коливається від 0,27 до 0,30 нм, а енергія зв'язку — від 8 до 29 кДж/моль. Такі зв'язки утворюються в димерах (Н2О)2, (HF)2 та ін. Для міцних зв'язків характерні коротші відстані (0,23... 0,26 нм) й високі значення енергії, близькі до енергій зв'язку в двохатомних молекулах (120... 250 кДж/моль). Міцний зв'язок реалізується в іоні [HF2]- де атом Гідрогену розміщений посередині між двома атомами Флюору (0,113 нм) і має дуже високу енергію (168 кДж/моль).
Енергія Н-зв'язку зменшується зі збільшенням температури, тому цей зв'язок характерніший для речовин у конденсованому стані.
3.Міжчасткові взаємодії. Вандерваальсова взаємодія молекул.
МІЖЧАСТИНКОВІ ВЗАЄМОДІЇ
Існування речовин у різних агрегатних станах свідчить про те, що між незарядженими частинками (атомами, молекулами) можуть діяти сили притягання, які не укладаються в звичайні валентні уявлення. Зокрема, дією цих сил пояснюються:
1) перехід з газуватого стану в рідкий або твердий;
2) відмінність між реальними та ідеальними газами. Сили взаємодії між частинками реальних газів уперше врахував Ван дер Ваальс (1873) у рівнянні стану газів;
3) явище Джоуля—Томсона (1854) — охолодження газу під час його адіабатного проникнення крізь пористу перетинку. Воно вказує на те, що при розширенні долаються сили притягання;
4) конденсація інертних газів, які не утворюють звичайних валентних зв'язків, у рідкий та твердий стани з виділенням енергії;
5) процеси адсорбції, сублімації, каталізу, розчинення і сольватації.
Ці сили являють собою взаємодію електронів і ядер різних частинок декількох типів, але їх об'єднують під загальною назвою міжчастпинкових, або вандерваальсових сил. Найважливішою і відмінною рисою вандерваальсових сил є їх універсальність, оскільки вони діють без винятку між усіма атомами і молекулами. Міжчастинкові сили відрізняються від хімічних тим, що вони виявляються на значно більших відстанях (на яких дія хімічних сил зникає), характеризуються малою енергією і відсутністю насичуваності та напрямленості. Вони швидко послаблюються зі збільшенням відстані між частинками, причому відштовхування спадає значно швидше притягання. Як правило, вандерваальсові сили між частинками зростають зі збільшенням числа електронів у частинці, тобто приблизно пропорційно формульній масі. Оскільки вони діють між валентно-насиченими частинками, то їх називають також залишковими силами.
ВАНДЕРВААЛЬСОВА ВЗАЄМОДІЯ МОЛЕКУЛ
Міжмолекулярна взаємодія має електромагнітну і квантово-механічну природу і складається з сил притягання та відштовхування. Сили притягання є результатом дії трьох ефектів: орієнтацій-ного, індукційного та дисперсійного:
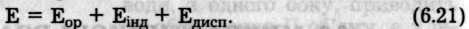
Енергія всіх трьох доданків пов'язана з дипольною взаємодією різного походження (рис. 6.47).
Орієнтаційний ефект виникає лише між полярними молекулами, які мають власний дипольний момент. Внаслідок хаотичного теплового руху молекули при зближенні орієнтуються так, що різнойменно заряджені кінці їхніх диполів притягаються (рис. 6.47, а). Енергія диполь-дипольного притягання визначається співвідношенням (Кеезом, 1912)
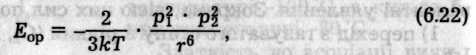
де р1 і р2 — постійні дипольні моменти взаємодіючих молекул; r — відстань між центрами диполів; k — константа Больцмана; Т — абсолютна температура.
Чим полярніші молекули, тим сильніше вони притягаються і тим більша енергія орієнтаційного ефекту. З підвищенням температури ефект зменшується, бо підсилений тепловий рух порушує взаємну орієнтацію диполів. Найсильніше орієнтаційний ефект виявляється у молекул, що мають великий дипольний момент і малі розміри (Н2О, NH3). У молекул, що мають малий дипольний момент (CO) або великі розміри (SO2), орієнтаційний ефект менший, а у неполярних частинок (СО2, інертні гази) він взагалі відсутній.
Індукційний ефект зумовлений дією індукованих диполів молекул, які можуть виникати в неполярних молекулах внаслідок постійного дипольного моменту сусідніх молекул (рис. 6.47, б). Взаємодія постійного диполя однієї молекули та індукованого диполя іншої спричинює зниження енергії системи з двох молекул. У свою чергу індукований диполь збільшує дипольний момент полярної молекули. Енергію взаємодії диполя з індукованим диполем обчислюють за формулою (Дебай, 1920)
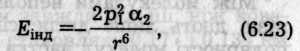
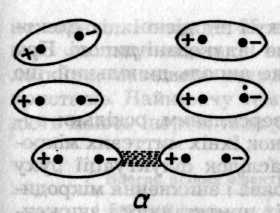

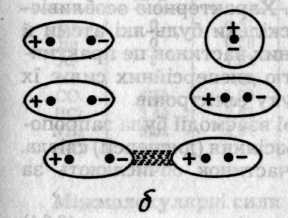
Рис. 6.47. Схематичне зображення орієнтаційного (а), індукційного (б)
та дисперсійного (в) ефектів між молекулами
Оскільки орієнтація наведеного диполя визначається лише напрямком постійного диполя, то енергія індукованої взаємодії не залежить від температури. Вона збільшується зі зростанням дипольного моменту й поляризованості та швидко спадає зі збільшенням відстані.
Енергія індукційного ефекту в десять-двадцять разів менша від енергії орієнтаційного ефекту. Вона суттєва лише для таких молекул, які легко поляризуються. Наприклад, індукційний ефект амоніаку вищий, ніж води, оскільки поляризованість молекул NH3 більша ніж Н2О, хоч дипольний момент амоніаку менший.
Під час взаємодії двох полярних молекул під дією їхніх електричних полів у них виникають додатково індуковані диполі. При цьому індукований ефект накладається на диполь-дипольний, що збільшує взаємне притягання.
Дисперсійний ефект є найбільш універсальним, оскільки зумовлюється взаємодією частинок за рахунок їхніх миттєвих мікро-диполів (рис. 6.47, в), які виникають внаслідок флуктуації руху електронів і коливань ядер. Синхронна поява і зникнення мікроди-полів різних частинок супроводжується їх притяганням і зниженням енергії системи. Якщо синхронність порушується, то диполі руйнуються й частинки відштовхуються. Характерною особливістю дисперсійних сил є їх загальність, оскільки будь-які атоми й молекули мають електрони. Для неполярних частинок це практично єдине джерело сил. Іншою особливістю дисперсійних сил є їх адитивність — результат узгодженого руху електронів.
Кількісна характеристика дисперсійної взаємодії була запропонована Лондоном (1930) на основі теорії розсіяння (дисперсії) світла. Енергію цієї взаємодії для однакових частинок обчислюють за формулою

де hvo/2 — енергія коливань атомів при Т = 0 К з частотою v0. Наближено величину hv0 можна вважати рівною потенціалу іонізації.
Відносний внесок кожного з розглянутих видів взаємодії у сумарний ефект залежить в основному від двох властивостей взаємодіючих молекул — полярності та поляризованості.
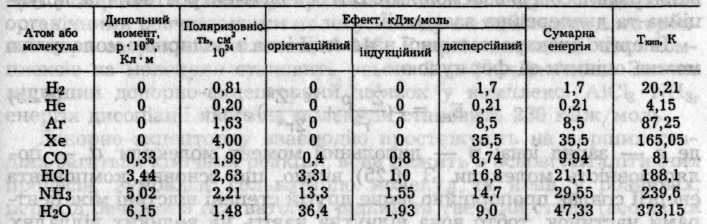
Між молекулами неполярних речовин або атомами інертних газів діють лише дисперсійні сили притягання. Зі збільшенням полярності молекул частка індукційних та орієнтаційних сил між ними зростає. Зі зростанням поляризованості молекул збільшується дисперсійний ефект. Індукційний ефект залежить від обох факторів, але сам має другорядне значення. Відносні значення трьох ефектів для деяких полярних і неполярних частинок наведені в табл. 6.2.
Знак мінус у рівняннях (6.22) — (6.24) означає, що для роз'єднання атомів або молекул необхідно виконати роботу, тобто, що діють сили притягання. Фактично ці рівняння стосуються енергії взаємодії. Силу притягання fпр визначають диференціюванням за r, a тому вона обернено пропорційна r7 у кожному випадку.
Порівняно з ковалентним зв'язком вандерваальсова взаємодія досить слабка. Про слабкість вандерваальсо-вого зв'язку свідчать низька температура плавлення, висока леткість, невелика твердість і незначна густина молекулярних кристалів. Найнижчу температуру плавлення й мінімальну твердість мають інертні гази у твердому стані.
Таблиця 6.2. Внесок різних ефектів в енергію вандерваальсової взаємодії
Міжмолекулярні сили принципово відрізняються від кулонівсь-ких своєю однозначністю — вони виявляються лише в притяганні. Однак при тісному зближенні частинок починають виявлятися сили взаємного відштовхування електронних оболонок атомів, що входять до складу частинок. Ці сили більш близькодіючі, ніж сили притягання, і зменшуються при збільшенні відстані за законом
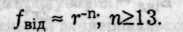
Внаслідок урівноваження сил притягання та відштовхування кожному атому й кожній молекулі можна приписати певний радіус. Природно, що вандерваальсові радіуси завжди більші, ніж ковалентні. Наприклад, для Іоду ці радіуси становлять 0,177 і 0,134 нм.
Міжмолекулярна взаємодія характеризується потенціальною енергією взаємодії, що враховує як енергію притягання, так і енергію відштовхування
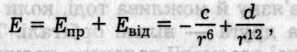
де с і d — сталі притягання та відштовхування. Обчислити потенціальну енергію при величезній кількості пар взаємодіючих частинок практично неможливо. Тому звичайно вираз Е(r) підбирають емпірично так, щоб він узгоджувався з експериментом. Особливо широкого застосування набула формула Леннард-Джонса (1924)
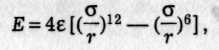
де ε — максимальна енергія притягання (аби глибина потенціальної ями.
Самостійна робота 4
Комплексні сполуки
Ко́мплексні сполу́ки або координаці́йні сполу́ки — складні хімічні сполуки, в яких можна виділити центральний атом (комплексоутворювач) і безпосередньо зв'язані з ним молекули абойони — так звані ліганди або аденти. Центральний атом та ліганди утворюють внутрішню сферу (комплекс); молекули або йони, які оточують комплекс — зовнішню координаційну сферу. Центральним атомом можуть бути як метали змінної валентності з позитивним зарядом ядра, так і неметали. Утворення комплексних сполук широко використовується в різноманітних галузях хімічної технології (виділення, очищення, розділення платинових, рідкісноземельних та деяких інших металів).
Комплексні сполуки — це сполуки, до складу яких входять комплексні частинки (комплекси), що вміщують центральний атом (комплексоутворювач) оточений лігандами. Утворення комплексів можна представити як результат взаємодії за донорно-акцепторним механізмом стабільних при звичайних умовах частинок: атомів, йонів або молекул.
Приведене вище визначення вказує, що, наприклад, такі частинки як CH4, SO2-4, NO-3 та інші недоцільно представляти як комплекси, оскільки частинки C+ і H-, S6+ і О2-, N5+ і О2- при звичайних умовах не існують.
Найхарактернішим комплексоутворення є саме для d-елементів.
Наявність значної кількості валентних частково заповнених орбіталей і схильність до утворення ковалентного зв'язку визначає значну кількість комплексних сполук, що утворюють d-елементи.
2.Комплексоутворюючі і ліганди
Ліганд (від лат. ligo — зв'язую) або адент в координаційній хімії — атом або група, безпосередньо зв'язана з одним або декількома центральними (комплексоутворювальними) атомами металу в комплексній сполуці.[1]
3.Номенклатура і класифікація комплексних сполук
Класифікація комплексних сполук.
Через численну кількість і різноманітність комплексних сполук важко провести єдину класифікацію. Існують класифікації комплексних сполук:
· По координаційному числу;
· По ступеню окиснення;
· По типу донорних атомів лігандів;
· По типу або природі координаційного зв’язку;
· По електронній конфігурації атома або іону металу.
Широко прийнятою є класифікація комплексних сполук по типу лігандів, які утворюють внутрішню координаційну сферу комплексів.
Аміакати – комплекси, в яких лігандами є молекули аміаку, наприклад:;;. Відомі комплекси, аналогічні аміакатам, у який роль ліганду виконують молекули амінів: СН3NH2 (метиламін), C2H5NH2 (етиламін), NH2CH2CH2NH2(етилендиамін Еn). Такі комплекси називають амінатами.
Аквакомплекси – лігандом виступає вода:;;. Гідратовані катіони, які є у одному розчині, містять як центральну ланку аквакомплекс. У кристалічному стані деякі з аквакомплексів утримують і кристалізаційну воду, наприклад:;. Кристалізаційна вода не входить до складу внутрішньої сфери, вона зв’язана не так міцно, як координована, і легше віддається при нагріванні.
Ацидокомплекси – лігандами є аніони. До них належать комплекси типу подвійних солей, наприклад, (їх можна подати як продукт поєднання двох солей – PtCl4 × 2KCl, Fe(CN)2 × 4KCN); комплексні кислоти H2[SiF6], H2[CoCl4]; гідроксокомплексиNa2[Sn(OH)4], Na2[Sn(OH)6].
Різнолігандні. Між цими трьома класами існують перехідні ряди, які включають комплекси з різними лігандами: [Pt(NH3)4]Cl2,[Pt(NH3)3Cl]Cl, [Pt(NH3)2Cl2], K[Pt(NH3)3Cl3], K2[PtCl4].
Циклічні або хелатні (клішнеподібні) комплексні сполуки. Вони містять бі- або полідентатний ліганд
До групи хелатів належать і внутрішньокомплексні сполуки, у яких центральний атом входить до складу циклу, утворюючи ковалентні зв’язки з лігандами різними способами: донорно-акцепторними і за рахунок неспарених атомних електронів. Комплекси такого роду дуже характерні для амінокарбонових кислот. Найпростіший їх представник – амінооцтова кислота (гліцин) утворює хелати з іонами Cu2+, Pt2+, Rh3+.
Відомі також комплекси з складнішими амінокарбоновими кислотами та їхніми аналогами. Такі ліганди називають комплексонами.
Хелатні сполуки мають особливу міцність, оскільки центральний атом у них ніби “блокований” циклічним лігандом. Найбільшу стійкість мають з п’яти- і шестичленними циклами. Комплексони так міцно зв’язують катіони металу, що при добавлянні їх розчиняються такі погано розчинні речовини, як сульфати кальцію і барію, оксалати і карбонати кальцію. Тому їх застосовують для пом’якшення води, маскування “зайвих” іонів металу при фарбуванні і виготовленні кольорової плівки. Широко застосовуються вони в аналітичній хімії.
Багато органічних лігандів хелатного типу є дуже чутливими і специфічними реагентами на катіони перехідних металів. До них належать, наприклад, диметилглікосим, запропонований Л.О.Чугаєвим, як реактив на іони Ni2+ та Pd2+.
Велику роль відіграють хелатні сполуки і в природі. Всі перелічені класи комплексних сполук є одноядерними, бо мають один центральний атом. Трапляються комплекси і складнішої структури, які містять два або кілька центральних атомів одного і того самого або й різних елементів. Ці комплекси називаються полі (багато) ядерними.
Місткові – комплекси з містковими атомами або групами атомів, наприклад з містковими атомами хлору (хлоро-), (оксо-), (аміно-), (гідроксо-).
[Сr(NH3)5 – OH ® (NH3)5Cr]Cl3 біядерний з однією містковою групою ОН.
Al2Cl6 біядерний з двома містковими хлоро- групами – Cl ®.
Кластери – комплекси, в яких атоми металу безпосередньо зв’язані один з одним. Це спостерігається наприклад, в димерах[(СО)5Mn–Mn(CO)5], [Re2H2Cl8]2-.
До багатоядерних сполук належать також ізополі- і гетерополікислоти.
|
|
Дата добавления: 2015-05-23; Просмотров: 2618; Нарушение авторских прав?; Мы поможем в написании вашей работы!