КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Работа с проектами Khimera
 |
Главный Khimera, работающий объект, является проектом. Работа с программой выполнена, главным образом, посредством управления с проектами через различные диалоговые окна, которое открывается из главного меню или панели инструментов.
Есть несколько типов проектов Khimera - проекты для вычисления элементарных особенностей процесса или реакции и проекты для особенности вещества и смеси вычисления:
• Проект реакции - для элементарного темпа процесса или вычисления поперечного сечения
• Проект вещества – для вещества термодинамическое имущественное вычисление
• Проект электронных состояний – для вычисления термодинамических свойств вещества, принимающего во внимание электронное возбуждение
• Проект смеси – для вычисления транспортных свойств смеси
Соответственно, режим функционирования программы и доступных команд строки меню и кнопок зависит от проекта, в настоящее время работают под. Проекты реакции используют меню реакции и вещество; вещество и проекты смеси используют только меню смеси и вещество
4.3.1 Создание нового проекта реакции
Создать новый проект реакции:
1. Выберите Файл → Новая опция меню
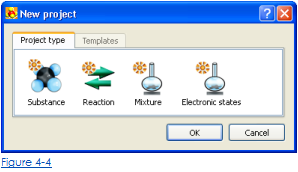
2. В типе Проекта диалог выбирают Реакцию в качестве типа проекта (рис. 4-4). Диалог для входа в уравнение реакции появится
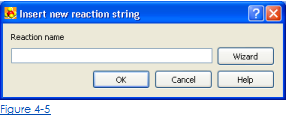
3. Войдите в уравнение реакции в область имени Реакции (Рис. 4-5) и нажмите Ok.
Новый проект будет создан, и диалог исследователя Реакции с новой реакцией будет открыт. Теперь Вы готовы выбрать модель для вычисления и ввести обязательные параметры.
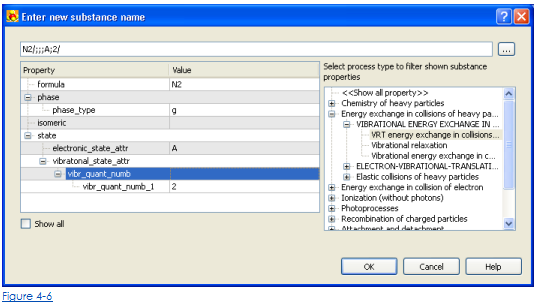
Чтобы упростить ввод правильного имени вещества для реактивов, возможно использовать волшебника синтаксиса имени вещества (рис. 4-5). Волшебник может быть открыт, щелкая кнопкой Wizard во Вставке новый диалог реакции (рис. 4-6). Волшебник синтаксиса имени вещества позволяет выбирать класс процессов и выбирать параметры имени вещества, которые должны быть определены. После спецификации обязательных параметров череда имени сущностей будет сформирована и вставлена в последовательность имени реакции. Для получения дополнительной информации о синтаксисе имени Вещества посмотрите Синтаксис для Веществ и Реакций в KHIMERA).
|
|
|
4.3.2 Создание нового проекта вещества
Создать новый проект вещества:
1. Выберите Файл → Новая опция меню
2. В типе Проекта диалог выбирают Вещество в качестве типа проекта (рис. 4-4). Диалог для ввода имени вещества появится.
3. Введите имя вещества и нажмите Ok.
Новый проект будет создан, и диалог исследователя Вещества с будет открыт. Теперь Вы готовы выбрать модель для вычисления и ввести обязательные параметры.
4.3.3 Открытие существующего проекта
Открыть существующий проект Khimera:
1. Выберите Файл → Открытая опция меню.
2. Выберите файл проекта в диалоге браузера файла и нажмите Open.
Проект будет загружен в программу. Если текущий проект не был сохранен, диалоговое окно открывает предоставление пользователю возможность сохранить проект.
4.3.4 Экономия проектов
Чтобы сохранить проект, использование Регистрирует/Экономит или Регистрирует/Экономит как опции меню. Если проект еще не был сохранен, имя и весь путь к справочнику сохраняют проект, будет требоваться. Уже чтобы сохранить сохраненный проект с другим именем или путем Регистрируют/Экономят, поскольку выбор должен использоваться.
4.4 Работа с веществами
4.4.1 Вещества в Khimera
Для каждого вещества, участвующие в Khimera, предполагают, что объект вещества, содержащий много параметров, характеризующих состояние сущности и свойства, создан. Вещества в Khimera могут представить как независимый проект или как часть реакции или проекта смеси вещества. Работа с отдельным веществом не отличается по зависимости от типа проекта Khimera, которые содержат это вещество. После того, как проект вещества создан, или вещество включено в проект реакции, параметры вещества могут быть рассмотрены и отредактированы через окно исследователя Вещества (Рис. 4-7), который может быть открыт, выбирая Вещество → опция меню Исследователя, если вещество является независимым проектом или использованием окна Исследователя Реакции, щелкая одной из кнопок с названиями веществ, участвующих в реакции.
|
|
|
4.4.2 Определение параметров вещества
Параметры вещества могут быть получены из файлов продукции программ квантовой химии или от базы данных вещества Khimera; они также могут быть введены вручную посредством диалога Исследователя Вещества. Есть много параметров вещества, которые могут быть отредактированы через Исследователя Вещества, но только некоторые из них требуются для вычисления с особой моделью Khimera. Эти параметры выдвинуты на первый план, если модель вычисления для проекта выбрана. Ниже, есть описание параметров вещества и способ его модификации через Исследователя Вещества.
4.4.2.1Entering молекулярные параметры геометрии (молекулярная структура, газовый кинетический радиус,
молярная масса, симметрия и момент инерции)
Определить параметры, характеризующие геометрию молекулы:
• геометрическая структура вещества,
• массы атомов
• вещество молекулярная масса
• симметрия молекулы
• газовый кинетический радиус
• моменты инерции.
Выберите вкладку Geometry (рис. 4-7) Исследователя Вещества
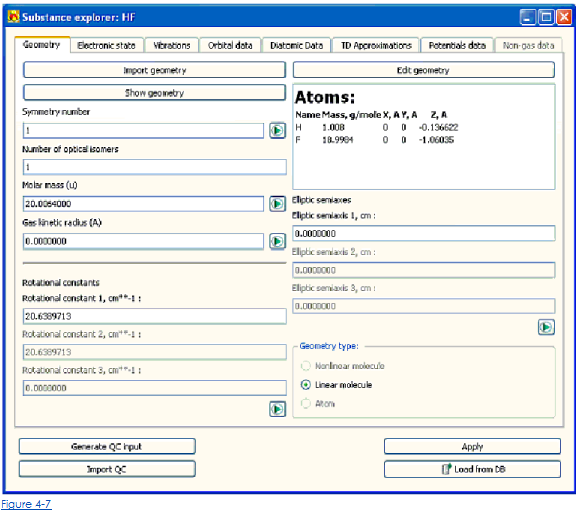
По умолчанию программа автоматически импортирует часть этого эти параметры от базы данных.
• Чтобы загрузить параметры геометрии от файла, содержащего результаты вычисления квантовой химии, щелкните кнопкой Import Geometry.
• Чтобы исправить или войти в молекулярные данные о геометрии вручную, нажмите кнопку геометрии Edit. Это открывает редактора для молекулярных параметров геометрии (Рис. 4-8). Здесь пользователь может добавить или удалить атомы, составляющие молекулу, и определить их массы и координаты.
• Чтобы установить массы атомов к средним ценностям массы изотопа от периодической таблицы нажимают кнопку масс Reset.
|
|
|
• Чтобы восстановить имя атомов и массы с имени вещества нажимают Rebuild от кнопки имени
• Чтобы очистить все молекулярные данные о геометрии щелкают кнопкой Clear All.
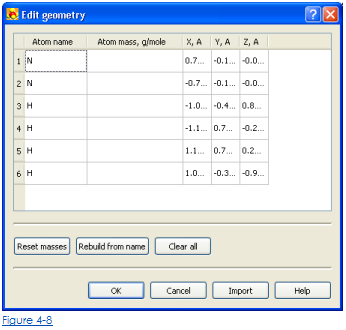
• Войти или изменить
• число симметрии,
• молярная масса,
• молекулярный радиус,
• постоянные вращения (взаимный момент инерции) для трех топоров (для нелинейной молекулы)
Нужно войти или отредактировать ценности в соответствующих полях ввода вкладки Geometry (рис. 4-7).
Возможно вычислить эти ценности от данных по координатам атомов и массам. Сделайте это, щелкая кнопкой , расположенной справа или выше соответствующего поля ввода.
• Определить тип геометрии молекул (линейный, нелинейный, атом), если это не определено автоматически, избранная соответствующая стоимость от типа Геометрии радио-кнопки.
• Чтобы визуализировать структуру геометрии молекулы нажимают кнопку геометрии Show.
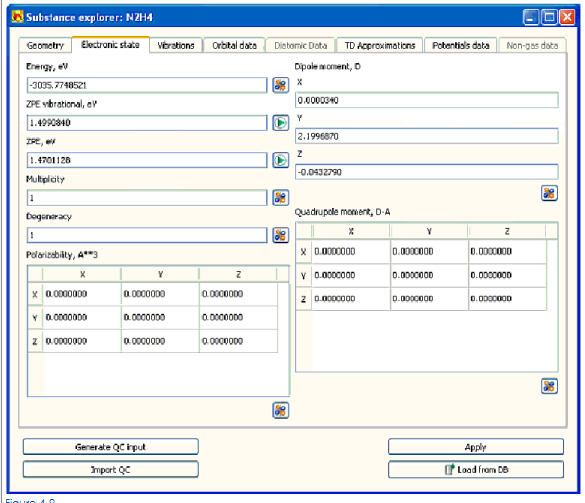
4.4.2.2Entering параметры электронных состояний молекул
Войти или отредактировать параметры электронного состояния вещества:
• энергия, eV
• энергия нулевых колебаний,
• разнообразие,
• вырождение,
• поляризуемость,
• дипольный момент
• момент четырехполюсника
выберите вкладку Electronic State (Рис. 4-9) и отредактируйте ценности в соответствующих полях ввода или столах.
Возможно вычислить ZPE и вибрационные ценности ZPE от частот молекулярного колебания и параметров внутреннего вращения. Чтобы сделать это щелкают кнопкой  справа от этих полей ввода.
справа от этих полей ввода.
• Загрузить ценность параметра от файла, содержащего вычисление квантовой химии, заканчиваются кнопка  щелчка справа или прореветь соответствующий поданный вход.
щелчка справа или прореветь соответствующий поданный вход.
4.4.2.3Entering параметры на молекулярных колебаниях и внутренних вращениях
Определить параметры молекулярных колебаний:
• Частоты молекулярных колебаний
• Параметры внутреннего вращения
выберите вкладку Vibrations (Рис. 4-10).
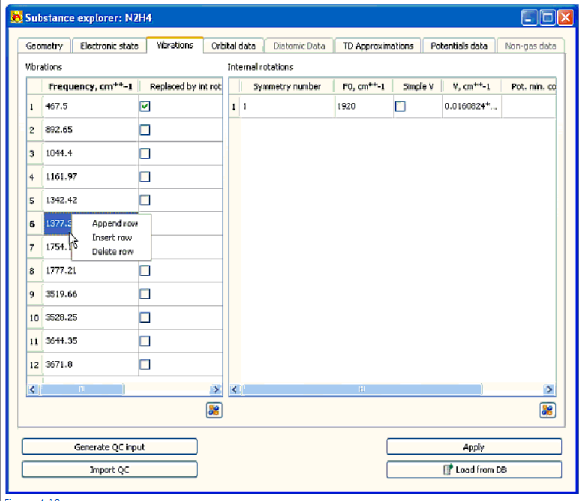
Вкладка Vibrations содержит стол со списком вибрационных частот и стол с параметрами внутренних вращений.
|
|
|
• Чтобы войти или отредактировать ценность вибрационной частоты - редактируют соответствующую стоимость в столе Vibrations. Используйте контекстное меню, появившееся по правильному щелчку кнопки мыши, чтобы добавить и удалить ряды за столом.
• Отметить частоту, замененную внутренним вращением (для случая, когда вибрационные частоты вычислены пакетом квантовой химии в гармоническом приближении и параметрах внутреннего вращения, вычислены независимо), избранный Замененный во флажке гнили в соответствующей строке таблицы.
• Загрузить параметры от файла с результатами вычисления квантовой химии –
щелкните кнопкой .
Чтобы определить параметры внутреннего вращения используют стол Внутренних вращений. Каждый ряд стола содержит параметры особого внутреннего вращающего устройства (все вращающие устройства считаются как независимые):
• Число симметрии - определяет число эквивалентных положений вращающего устройства за поворот на 360 градусов.
• F0, cm-1 кинетический фактор - представляет интересы как постоянная вращения вращения в целом и равен отношению кинетической энергии вращения к брусковой частоте вращения. Для сложных случаев, e. g., когда движение вращающего устройства включает колебательную часть, кинетический фактор может быть определен как функция ориентации вращающего устройства; это определено тогда с точки зрения коэффициентов расширения Фурье. В большинстве случаев угловая зависимость является низкой, таким образом, соответствующие коэффициенты Фурье должны быть маленькими по сравнению с постоянным сроком расширения.
• Простой V флажков – определяют путь, как гармоническая функция вращения определена (см., что описание ревет).
• V, cm-1 – гармоническая функция V, который описывает зависимость потенциальной энергии на ориентации вращающего устройства. Для точных вычислений гармоническая функция определена как последовательное расширение Фурье. Постоянный срок расширения является ориентиром и не затрагивает результатов вычисления. В простом случае (Простой V выбранных опций) гармоническая функция может быть описана двумя параметрами: число потенциальных минимумов определено от формы вещества и глубины потенциала, хорошо оценен как разность энергий между положениями вращающего устройства с максимальной и минимальной энергией.
• Количество Pot.min. – число минимумов гармонической функции. Используется, если опция Simple V выбрана.
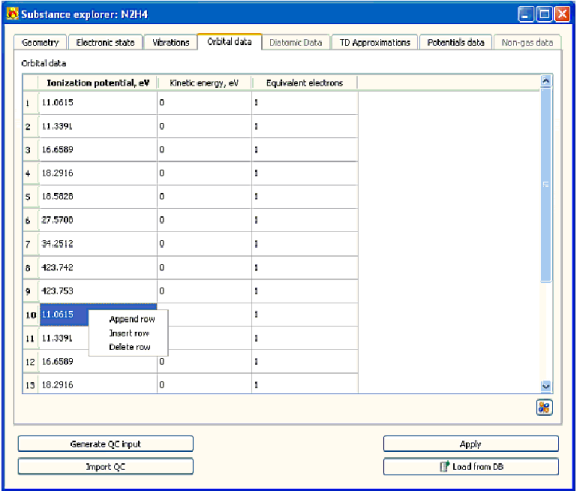
4.4.2.4Entering параметры электрона orbitals
Определить электронные orbitals параметры
• ионизационный потенциал
• кинетическая энергия
• число электронов на орбитальном
выберите Орбитальный счет данных (Рис. 4-11).
Счет состоит в столе, в котором ряд соответствует особый электронный орбитальный.
• Чтобы войти или отредактировать параметры электронного orbitals – редактируют соответствующие ценности в столе. Используйте контекстное меню, активированное, щелкая правой кнопкой мыши по мыши на столе, чтобы добавить, вставить, и удалить ряд из стола
• Чтобы загрузить электронные orbitals параметры от файла с результатами вычисления квантовой химии – щелкают кнопкой .
4.4.2.5Entering параметры для двухатомных молекул
Определить параметры двухатомных молекул:
• атомные параметры потенциала взаимодействия (молекулярный термин)
• параметры микроструктуры молекулы
выберите Двухатомный счет данных.
Список параметров, характеризующих термин двухатомной молекулы, зависит от государственной природы (стабильный или нестабильный. Чтобы определить термин тип выбирают соответствующий выбор (связанный – для стабильного, разложение - для нестабильного) от термина тип роняют список.
• Чтобы определить величину проектирования углового орбитального импульса на молекулярной оси входят в него в поданном входе Лямбды.
Потенциал взаимодействия атомов в двухатомной молекуле может быть введен как зависимость энергии на расстоянии между атомами (в табличной форме) или с параметрами приближения.
• Чтобы выбрать способ спецификации потенциала выбирают выбор Стола или Приближения в радио-кнопке.
Рисунок 4-12 показывает представление о Двухатомном счете данных, когда способ Стола для Связанного терма отобран.
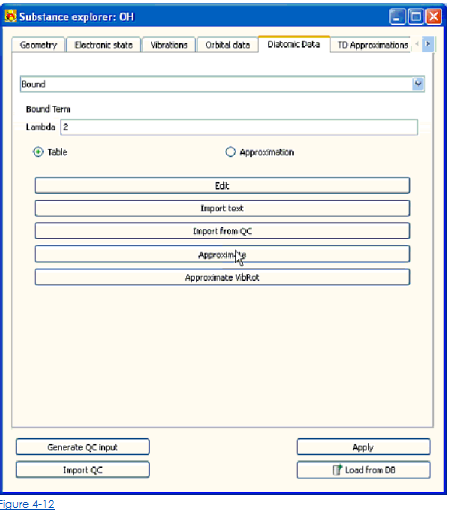
• Войти или отредактировать атомный потенциал взаимодействия вручную в табличной форме:
1. Щелкните кнопкой Edit.
2. При открытом диалоге со столом входят или редактируют межатомное расстояние и переписывался стоимость энергии взаимодействия.
• Чтобы импортировать двухатомные потенциальные данные из текстового файла нажимают текстовую кнопку Import
• Чтобы импортировать двухатомный потенциал из результатов вычисления квантовой химии нажимают Import from QC. Посмотрите, что Импорт формирует королевского адвоката для подробного описания процедуры импорта.
Данные по двухатомному потенциалу взаимодействия используются моделями вычисления в форме коэффициентов приближения. Приближение двухатомных потенциальных данных, вошедших в табличную форму или импортированных из результатов вычисления квантовой химии, выполнено Khimera автоматически. Но есть возможность приблизить его вручную, чтобы проверить или изменить данные, переданные моделям вычисления. Если есть данные по двухатомному потенциалу и в столе и в форме приближения, присутствуют, данные в форме приближения используются для вычисления, и автоматическое приближение не выполнено.
Возможно приблизить потенциал взаимодействия Морзе или Вибрационно-вращательными формулами (для Связанного терма) или показательной функцией (для термина разложения).
• Чтобы приблизить двухатомный потенциал формулой Морзе (для Связанного терма) или показательной функцией (для термина разложения) щелкают кнопкой Approximate.
• Чтобы приблизить двухатомный потенциал вибрационно-вращательными формулами – щелкают кнопкой Approximate VibRot.
Рассмотреть вычисленный выключатель параметров приближения к способу Приближения.
Спецификация параметров приближения для Связанного терма
Когда способ Приближения выбран для спецификации атомного потенциала, взгляды диалога как показано на рисунке. 4-13 (для Связанного терма).
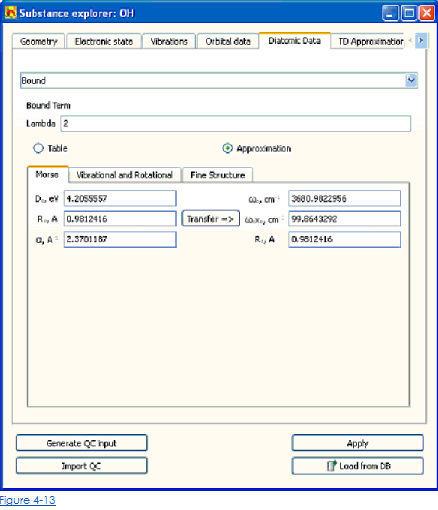
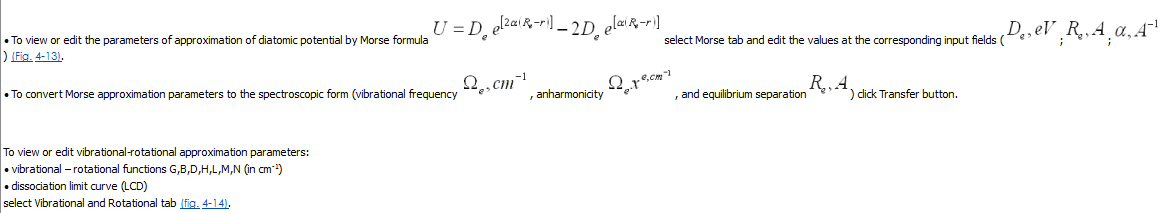
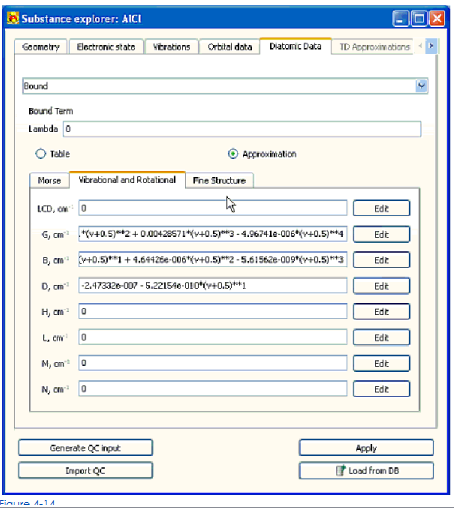
Вибрационно-вращательные функции определены в форме многочленной функции, зависящей от иррационального числа. Жидкокристаллическая функция – в форме многочленной функции, зависящей от вращательного числа.
Отредактировать коэффициенты полиномиалов вибрационно-вращательных функций и ЖК-монитора
1. Щелкните кнопкой Edit, расположенной справа от соответствующего поданного входа
2. В открытом степенном ряду редактор определяет коэффициенты полиномиалов и нажимает Ok. Добавить или удалить компонент многочленного контекстного меню использования появились по правильному щелчку кнопки мыши.
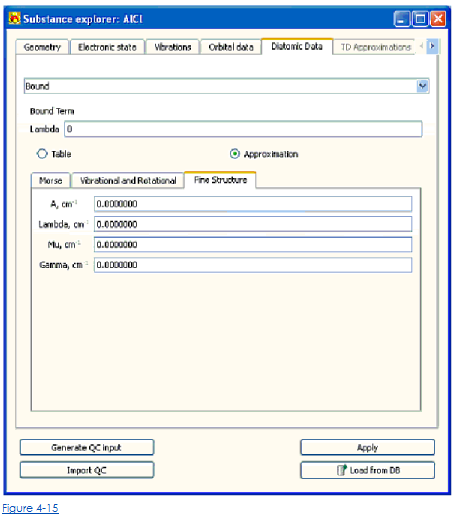
Определить параметры микроструктуры двухатомной молекулы
• A
• Лямбда
• Му
• Гамма
Выберите счет Микроструктуры (Рис. 4-15) и отредактируйте ценности в соответствующих полях ввода.

4.4.2.3Entering или вычисление параметров приближения термодинамических функций
Для приближения термодинамических функций, таких как теплоемкость, энтропия, теплосодержание и свободная энергия Гиббса, программа использует формулы IVTAN, которые позволяют этим ценностям быть полученными как функции температуры, используя обеспеченные полиномиалы (см. Вычисление термодинамических функций).
Коэффициенты для этих полиномиалов могут быть вычислены основанные на молекулярных данных о геометрии, импортировали из базы данных или входили вручную. Спецификация коэффициентов приближения термодинамических функций, требуемых в особенности для транспортного имущественного вычисления.
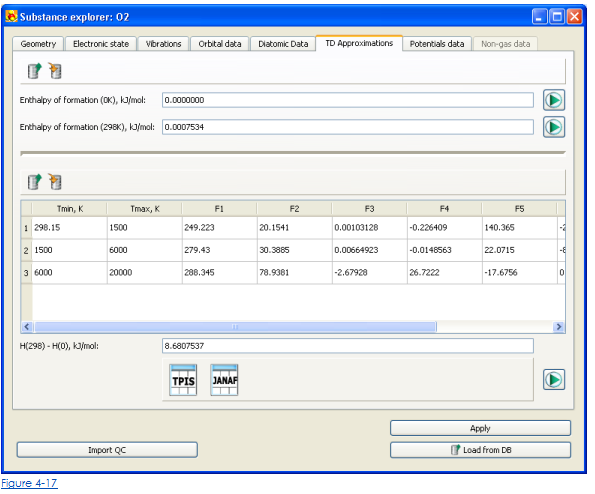
Отредактировать или вычислить
• Многочленные коэффициенты для приближения термодинамических функций
• Теплосодержание вещества формирования
выберите вкладку TD Approximations (Fig.4-17).
Вход или импортирование многочленных коэффициентов и теплосодержания формирования
Многочленные коэффициенты приближения термодинамических функций представлены в столе с корреспондентом рядов к особому температурному интервалу.
• Чтобы войти или отредактировать многочленные коэффициенты - редактируют соответствующие ценности в клетке стола. Чтобы добавить или удалить ряд - используют контекстное меню, которое может быть открыто правильным щелчком кнопки мыши.
• Чтобы войти или изменить теплосодержание формирования вещества – входят или редактируют стоимость в Теплосодержании формирования (298K) поданный.
• Чтобы загрузить содействующие ценности приближения от базы данных щелкают кнопкой  , расположенной выше стола с коэффициентами.
, расположенной выше стола с коэффициентами.
Чтобы загрузить ценности теплосодержания формирования от базы данных щелкают кнопкой , расположенной выше группы областей теплосодержания формирования.
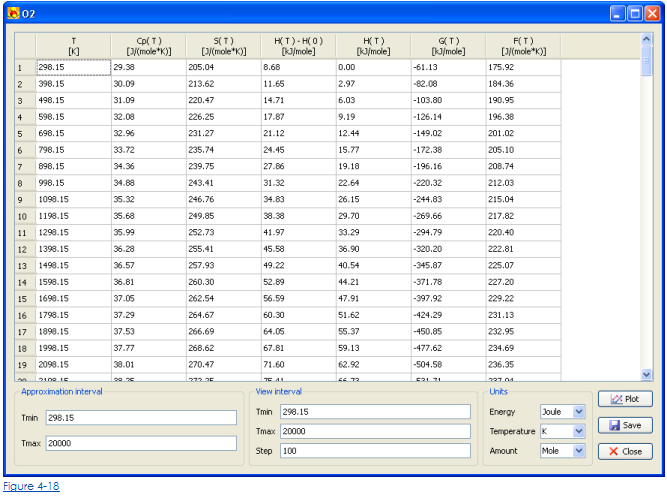
Спецификация теплосодержания формирования вещества и коэффициентов полиномиалов приближения достаточно для вычисления термодинамических функций. Khimera способен, чтобы представлять температурную зависимость термодинамических функций в форме TPIS и столов JANAF.
• Чтобы открыть TPIS или стол JANAF с зависимостью температуры термодинамической функции щелкают кнопкой TPIS или JANAF соответственно. Диалог, который содержит TPIS или стол JABAF, появится (Рис. 4-18).
• Чтобы подготовить вычисленную зависимость температуры термодинамической функции щелкают кнопкой Plot и выбрать функцию, чтобы составить заговор (Рис. 4-19)
Вычисление коэффициентов приближения термодинамических функций
Зависимость температуры термодинамических функций и соответствующие коэффициенты полиномиалов могут быть вычислены, используя данные по молекулярной геометрии, вибрационным спектрам, параметрам внутреннего вращения и электронным orbitals данным. Эти данные должны быть определены, используя соответствующие коробки с исследователем вещества.
Чтобы начать термодинамические функции и содействующие вычисления полиномиалов, нужно осуществить следующее:
1. Войдите или импортируйте из результатов вычисления квантовой химии данные по геометрии, вибрационным частотам и параметрам внутреннего вращения (если таковые имеются) и особенности электронного состояния, используя соответствующие коробки с исследователем Вещества
2. Выберите счета приближения TD
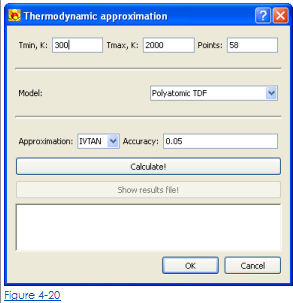
3. Нажмите  ревут стол с коэффициентами полиномиалов.
ревут стол с коэффициентами полиномиалов.
4. В открытом (Рис. 4-20) определяют температурный интервал и число очков для вычисления, выбирают, модель вычисления от снижения вниз перечисляют и определяют точность приближения.
5. Щелкните кнопкой Calculate.
6. Щелкните Кнопкой ОК, чтобы добавить вычисленные коэффициенты полиномиалов к столу в счете приближения TD.
Вычисление теплосодержания формирования вещества
Khimera способен, чтобы вычислить теплосодержание формирования вещества, основанное на данных по ценностям молекулярных и атомных энергий. Чтобы вычислить теплосодержание формирования вещества, нужно осуществить следующие шаги:
1. Импортируйте ценность энергии молекулы от файла с результатами вычисления квантовой химии (Гауссовский подход Gn) использование кнопки QC импорта исследователя вещества.
2. Щелкните кнопкой  справа от “Теплосодержания формирования, (0k)” область, чтобы открыть диалог для спецификации параметров атома
справа от “Теплосодержания формирования, (0k)” область, чтобы открыть диалог для спецификации параметров атома
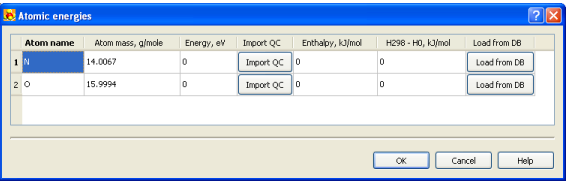
3. При открытом диалоге определяют
• Энергия атома (может быть импортирован из результатов вычисления квантовой химии, нажимая кнопку Import QC),
• Теплосодержание формирования атома и изменение теплосодержания (H298-Ho) могут быть импортированы из базы данных, нажимая Груз от кнопки DB.
4. Нажмите Хорошо, чтобы закрыть диалог и вычислить теплосодержание формирования вещества в 0K.
5. Если Вам вычислили коэффициенты приближения термодинамических функций или импортированный из базы данных, Вы можете вычислить теплосодержание формирования вещества в 298 K, нажимая кнопку  справа от “Теплосодержания формирования, (298K)” область.
справа от “Теплосодержания формирования, (298K)” область.
4.4.2.6Entering данные о потенциале межмолекулярного взаимодействия
Кимера использует потенциалы межмолекулярного взаимодействия для имущественных вычислений транспортировки смеси;
Khimera поддерживает следующее приближение для межмолекулярного потенциала:
• Леннард-Джонс, (12-6),
• Stockmayer,
• Леннард-Джонс (m6),
• Родившийся-Mayer,
• Букингемский угол и
• HFD-B (Азиз),
• pointwise (С начала) потенциал
• Чтобы определить параметры потенциала межмолекулярного взаимодействия для химически чистого вещества выбирают Потенциальный счет данных и входят в параметры приближения в соответствующие поля ввода. Выберите Использование во флажке вычисления для потенциального приближения, которое Вы хотите использовать в вычислении.
• Чтобы войти с начала в потенциальную прессу данных Редактируют кнопку функции и вводят ценности в столе. Вы можете импортировать с начала потенциальные данные из текстового файла, нажимая кнопку Import.
• Чтобы импортировать межмолекулярные потенциальные данные из базы данных нажимают Груз от кнопки DB.
4.4.2.7 Вход в параметры веществ на поверхности
Определить параметры, требуемые для вычисления темпа газово-поверхностных процессов:
• Занятые места - число занятых вакансий
• Частота седла, cm-1 - обратная длина волны колебаний в потенциале седла (в перпендикуляре самолета к смещению от одной вакансии до другого.
• Расстояние места - среднее расстояние между вакансиями в ангстремах
• Плотность – поверхностная плотность вещества
Выберите Негазовый счет данных и определите ценности, требуемые для вычислений в соответствующих полях ввода.
|
|
|
|
|
Дата добавления: 2015-05-09; Просмотров: 292; Нарушение авторских прав?; Мы поможем в написании вашей работы!