КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Проводится набор в секцию 3 страница
|
|
|
|
Ограниченный (по спину) метод Хартри-Фока (ОХФ, англ. RHF) -метод самосогласованного решения уравнения Шредингера, в котором многоэлектронная волновая выбирается в виде единственного детерминанта Слейтера, построенного из пространственных орбиталей, занятых парой электронов с противоположными спинами. Используется для систем с открытой и закрытой оболочкой. См. также неограниченный метод Хартри-Фока (НХФ, англ. UHF).
Одноэлектронное приближение – модель, в которой движение каждого электрона предполагается происходящим в поле ядер и в усредненном поле остальных электронов.
Оператор -символ, обозначающий математическую операцию, с помощью которой из одной функции получается другая. Каждому оператору отвечает уравнение на собственные значения А f = af, a - в общем случае комплексное число, называемое собственным значением оператора А, f называется собственной функцией оператора А.
Оператор кинетической энергии молекулы - T = 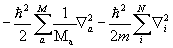 , где Мa- масса ядра a; m - масса электрона;
, где Мa- масса ядра a; m - масса электрона; 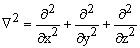 - оператор Лапласа (лапласиан). Дифференцирование ведется по координатам ядер Ra и по координатам электронов ri. Оператор описывает кинетическую энергию электронов и ядер.
- оператор Лапласа (лапласиан). Дифференцирование ведется по координатам ядер Ra и по координатам электронов ri. Оператор описывает кинетическую энергию электронов и ядер.
Оператор потенциальной энергии молекулы -
V = 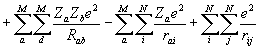 , где
, где 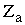 и
и 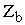 - атомный номер элемента, e - заряд электрона, Rab - расстояние между ядрами, rai - расстояние между ядрами и электронами, rij- между электронами, e0 – электрическая постоянная. Оператор описывает отталкивание ядер, притяжение электронов к ядрам и отталкивание электронов.
- атомный номер элемента, e - заряд электрона, Rab - расстояние между ядрами, rai - расстояние между ядрами и электронами, rij- между электронами, e0 – электрическая постоянная. Оператор описывает отталкивание ядер, притяжение электронов к ядрам и отталкивание электронов.
Оптимизация геометрии молекулы– поиск устойчивой конфигурации молекулы, отвечающей локальному или глобальному энергетическим минимумам.
Орбитали слейтеровского типа (ОСТ или STO) – функции, аппроксимирующие атомные орбитали. В сферических координатах 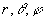 имеют вид
имеют вид 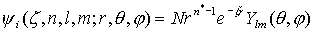 .Здесь
.Здесь 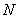 – нормировочный множитель,
– нормировочный множитель, 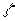 - орбитальная экспонента,
- орбитальная экспонента, 
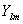 - сферическая гармоника, n, l, и m - квантовые числа. Орбитали слейтеровского типа правильно описывают асимптотическое поведение электронов как вблизи ядра, так и на больших расстояниях от него.
- сферическая гармоника, n, l, и m - квантовые числа. Орбитали слейтеровского типа правильно описывают асимптотическое поведение электронов как вблизи ядра, так и на больших расстояниях от него.
|
|
|
Орбитали гауссова типа (ОГТ или GTO) функции, аппроксимирующие атомные орбитали. В декартовых координатах x, y, и z имеют вид  , где – нормировочный,
, где – нормировочный, 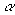 - орбитальный экспоненциальный множитель, r2= x2 + y2 + z2. Числа l и m определяют угловую часть ОГТ в декартовых координатах: их сумма (l+м+n) аналогична угловому квантовому числу для атомов и используется, чтобы обозначать функции s-типа (l=0), p-типа (l=1), d-типа (l=2), f-типа (l=3), и т.д. Строго говоря, декартовы ОГT не являются орбиталями: они лишь простые и удобные математические функции, которые часто называют гауссовыми примитивами.
- орбитальный экспоненциальный множитель, r2= x2 + y2 + z2. Числа l и m определяют угловую часть ОГТ в декартовых координатах: их сумма (l+м+n) аналогична угловому квантовому числу для атомов и используется, чтобы обозначать функции s-типа (l=0), p-типа (l=1), d-типа (l=2), f-типа (l=3), и т.д. Строго говоря, декартовы ОГT не являются орбиталями: они лишь простые и удобные математические функции, которые часто называют гауссовыми примитивами.
Ортогональные волновые функции – две функции ψ1 и ψ2 являются ортогональными, если интеграл S12= ∫dv ψ*1(r) ψ2(r) равен нулю. S12 называется интегралом перекрывания. Ортогональными являются собственные функции эрмитового оператора, соответствующие различным собственным значениям.
Особые точки скалярной функции – точки, в которых значения первых производных функции по аргументам равны нулю. Различают следующие типы особых точек: минимум, максимум, локальный минимум (а), седловая точка (б), максимум (в).
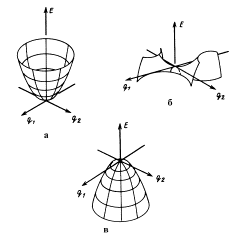
Особые точки поверхности потенциальной энергии – точки на поверхности потенциальной энергии (ППЭ), в которых выполняется условие стационарности: производная от потенциальной энергии U по всем нормальным колебательным координатам Qi (координат Qi, в базисе которых матрица Гессе диагональна) равна нулю: ¶U/¶Qi = 0, i = 1, 2,…, 3N–6(5). В минимуме, отвечающем равновесному состоянию системы, все собственные значения матрицы вторых производных потенциальной энергии по координатам (матрицы Гессе Нij=¶2 U/¶Qi¶Qj) положительны: Нii = ¶2U/¶Qi2 > 0. В седловой точке, отвечающей переходному состоянию - одномерном локальном максимуме - из нормальных координат одно и только одно из собственных чисел гессиана отрицательно. При квантово-химическом описании химических реакций предполагают, что реагентам, продуктам, пред- и послереакционным комплексам соответствуют локальные минимумы, а переходному состоянию – седловая точка ППЭ химической реакции.
|
|
|
Параметризация силового поля – подбор параметров для расчета потенциальных функций, определяющие так называемое силовое поле молекулы. Численное значение параметров выбирается так, чтобы получить согласие рассчитанных и экспериментальных характеристик молекулы. Параметрами являются равновесные межъядерные расстояния (длины связей) и валентные углы, а также силовые постоянные, то есть коэффициенты жесткости упругих сил, связывающих пары атомов. Простейшие модели молекулярной механики учитывают растяжения связей (Uраст.), деформацию валентных (Uдеф.) и двугранных (торсионных) углов (Uторс.), взаимодействие валентно несвязанных атомов, называемое также ван-дер-ваальсовым взаимодействием (Uвдв.), электростатические вклады (Uэл-стат.) и т.д.:
U = SUраст + SUдеф + SUторс + SUвдв + SUэл-стат.
Для каждого слагаемого записывается определенное аналитическое выражение (например, энергия электростатического вклада Uэл-стат описывается кулоновской функцией, возможно, с нецелыми зарядами на атомах в качестве параметров). Для описания потенциальной функции предельных углеводородов при умеренных требованиях к точности расчета достаточно около десяти параметров.
Правило 18 электронов - в устойчивых комплексах переходных металлов МLn общее число валентных электронов на связях M-L и несвязывающих молекулярных орбиталях центрального атома равно 18.
p-электронное приближение - при решении уравнений Рутана для ненасыщенных и ароматических молекул, чаще всего являющихся плоскими, s-АО считают неполяризованными и включают в атомный остов, а движение p-электронов рассматривают в потенциальном поле таких остовов. Волновая функция молекулы при этом представляется как произведение y = ys yp, где ys и yp - нормированные антисимметричные по отношению к s- и p-электронам функции, соответственно. Их можно разложить по слейтеровским детерминантам, составленных только из s- и только из p-МО. Волновая функция ys одинакова как для основного, так и для возбужденных состояний и все изменения связываются с p-электронами. Рассмотрение только p-электронов удовлетворяет вариационному принципу.
|
|
|
Поверхность потенциальной энергии – представление потенциальной энергии молекулярной системы как многомерной функции всех переменных (степеней свободы) в системе (например, длин связей и углов между ними). Потенциальная поверхность для молекулы с N атомами имеет 3N-6 независимых степеней свободы (три степени отвечают вращению и три - движению молекулярной системы). Важными особенностями потенциальной поверхности являются минимумы и седловые точки, которые представляют структуры переходных состояний, которые находят, оптимизируя геометрию молекул.
Полуэмпирические методы расчета – группа расчетных методов квантовой химии, использующих отказ от вычисления одноэлектронных и двуэлектронных интегралов, фигурирующих в методе ХФ. Вместо точного оператора Фока используется приближенный, элементы которого получают из эмпирических данных. Соответствующие параметры подбирают для каждого атома (иногда - с учетом конкретного окружения) и для пар атомов: таким образом, они являются либо фиксированными числами, либо зависят от расстояния между атомами.
Полуэмпирические методы намного быстрее, чем неэмпирические. Они применимы к большим (часто к очень большим, например - биологическим) системам и для некоторых классов соединений дают очень точные результаты. Однако это достигается за счет специально подобранных параметров, справедливых лишь в пределах узкого класса соединений. При переносе на другие классы, те же методы могут дать абсолютно неверные результаты. Кроме того, параметры расчета часто подбираются таким образом, чтобы воспроизводить те или иные молекулярные свойства, поэтому придавать физический смысл отдельным параметрам не следует.
|
|
|
Пренебрежение дифференциальным перекрыванием (NDO) – упрощение, применяемое в полуэмпирических квантово-механических расчетах. Считают, что из-за экспоненциального убывания АО двухэлектронными кулоновскими и обменными интегралами, содержащими произведения различных атомных орбиталей, зависящих от одного аргумента, можно пренебречь. Уменьшает число интегралов, описывающих в расчете взаимодействия электронов.
Полное пренебрежение дифференциальным перекрыванием (ППДП, англ. CNDO) - один из первых полуэмпирических методов. Используется для вычисления электронных свойств основного состояния систем с закрытой оболочкой, оптимизации геометрии и общей энергии. В настоящее время заменен более сложными полуэмпирическими квантово-химическими методами, такими как MNDO, AM1 и PM3.
Принцип сохранения орбитальной симметрии Вудворда-Хоффмана – носит общий характер и позволяет в ряде случаев предсказывать, будут ли соединения реагировать, по какому пути пойдет реакция и каких продуктов следует ожидать. Рассматриваются структура активных МО реагирующих веществ и знаки атомных орбиталей (АО), составляющих МО. Реакция называется разрешенной по симметрии, если в ходе ее симметрия активных МО (все участвующие в реакции занятые и вакантные МО реагентов и продуктов) сохраняется. Деление реакций на разрешенные и запрещенные по симметрии осуществляется с помощью корреляционных диаграмм. Корреляционную диаграмму взаимодействия между реагентами и продуктом строят простым соединением МО с одинаковой симметрией (S ® S и A ® A), учитывая, что разность энергий МО должна быть минимальна. Если это возможно и ни одна из соединяющих линий не пересекает нулевого уровня (правило непересечения), то реакция «разрешена» по симметрии.
Поляризационные функции – базисные функции, описывающие поляризацию орбиталей атомов соседями по молекуле. Состоят из функций с более высокими значениями орбитального квантового числа l, чем те, которые отвечают минимальному базису. p- и d-функции выступают как поляризационные функции для базисных функций для атомов с s электронами, d- и f- функции действуют как поляризационные функции для атомов с s и p электронами и д.т.
Порядок связи – мера простого, двойного или тройного характера связи. В теории молекулярных орбиталей в основу определения положен учет компенсации эффектов занятых электронами связывающих и разрыхляющих орбиталей (см. кратность связи по Герцбергу).
Постулат Хэммонда -связывает геометрию переходного состояния с тепловым эффектом реакции. По Хэммонду экзотермическим реакциям соответствует реагентоподобное переходное состояние, эндотермическим – продуктоподобное, теплонейтральным – лежащее примерно по середине между реагентами и продуктами.
Потенциал Букингема -основан на предположении об экспоненциальной зависимости сил отталкивания между молекулами от расстояния между ними. Используетсядля описания ван-дер-ваальсовых взаимодействий. Энергия взаимодействия между молекулами в этом случае зависит от расстояния R между ними: U(R) = be-aR – cR-6 – dR-8; a, b, c, d – постоянные. Это выражение справедливо только для неполярных сферически-симметричных молекул. Для расчета энергии взаимодействия более сложных систем в эмпирический потенциал вводят поправки. Чаще всего расчеты проводят с использованием упрощенного потенциала Букингема, в котором пренебрегают членом, включающим R-8, и рассматривают полное взаимодействие как сумму взаимодействий между всеми атомами подсистем.
Потенциал Леннарда-Джонса – служит для расчета энергии межмолекулярного взаимодействия. Основан на предположении о быстром возрастании сил отталкивания между молекулами на малых расстояниях, происходящем по закону R-n. Потенциал Леннарда-Джонса (6-12) получают при n = 12, зависимость R-6 в этом случае отражает наличие дисперсионных сил. Потенциальная энергия взаимодействия между системами не имеющими постоянных диполей описывается выражением U(r) = 4ε [(σ/r)12 - (σ/r)6], где ε и σ – эмпирические константы, r – расстояние между взаимодействующими системами; (σ/r)12- описывает отталкивание, (σ/r) 6 – описывает притяжение между системами.
Потенциал ионизации - энергия, которую необходимо сообщить системе, чтобы удалить какой-либо из ее электронов. По потенциалу ионизации можно судить о прочности связи электрона данной орбитали с атомным остовом.
Предел сходимости - вычисление методом ССП заканчивается, когда различие в энергии после двух последовательных итераций становится меньше, чем некая малая величина величина. Для полуэмпирических вычислений практический предел сходимости равен примерно 10-3 (по умолчанию 0,01 ккал/моль), для неэмпирических - 10-4-10 -5.
Приближение Борна-Оппенгеймера состоит в отделении движения ядер от электронного движения. Ядра намного тяжелее электронов и их можно считать неподвижными, рас-сматривая движение электронов относительно них. Уравнение Шредингера тогда может быть решено только для электронов при определенной ядерной конфигурации. Оправдано при квантово-химическом анализе поведения молекул в основном состоянии.
Приближение независимых частиц – наличие электрон-электронного отталкивания в гамильтониане не позволяет разделить координаты электронов и решить уравнение Шредингера аналитически для системы более двух электронов. Поэтому поведение каждого электрона описывается некоторой волновой функцией, подобно единственному электрону в атоме водорода, и зависит от поведения остальных электронов в среднем. В этом состоит суть приближения независимых частиц. Решения одноэлектронных уравнений – уравнений Хартри-Фока или Кона-Шэма – называются одноэлектронными волновыми функциями или орбиталями (в атоме - атомными орбиталями, в молекуле - молекулярными, в кристалле - кристаллическими). Для их расчета используется приближение самосогласованного поля.
Приближение самосогласованного поля (ССП) -в приближении независимых частиц действие всех остальных электронов на данный электрон заменяют средним полем, приближенно воспроизводящим их суммарное действие. Приближенная волновая функция (орбиталь) зависит от координат только рассматриваемого электрона: это дает возможность разделить переменные в уравнении Шредингера в сферической системе координат. Чтобы решить систему одноэлектронных уравнений с гамильтонианом, включающим усредненное межэлектронное взаимодействие, задают некоторый набор одноэлектронных функций, максимально близких к правильным, и с их помощью строят оператор, учитывающий отталкивание электронов. Затем решают набор одноэлектронных уравнений, возникающий из условия минимума среднего значения гамильтониана, вычисляемого с одноэлектронной волновой функцией. Полученные решения используют, чтобы построить "исправленный" оператор, вновь решают ту же систему уравнений, но теперь – с новым гамильтонианом и т.д. Процесс продолжается, пока получаемые собственные значения одноэлектронных энергий будут отличаться от полученных на предыдущей итерации лишь на очень незначительную величину. Этот процесс называется самосогласованием, а результирующее поле, создающее усредненный потенциал, называется самосогласованным полем.
Приближение центрального поля. Потенциал межэлектронного взаимодействия только в частных случаях (положительные одноэлектронные ионы, атомы инертных газов, атомы N, P и т.д.) сферически симметричен. Однако учет асферичности не улучшает заметно результат расчета, поэтому обычно используют усредненный по всем направлениям атомный потенциал, интегрируя его по углам. Вводимое таким образом приближение центрального поля позволяет рассматривать ССП-решения для любого атома как модифицированные решения для одноэлектронного водородоподобного атома. В этом случае потенциальная энергия зависит только от расстояния до ядра, т.е. сила притяжения к ядру носит центральный характер. Тогда переменные в уравнении Шредингера в сферических координатах разделяются и волновые функции, описывающие состояния электронов атома в пространстве (атомные орбитали), можно получить в аналитическом виде.
Растяжение связи - в эмпирической теории силового поля описывает изменение молекулярной потенциальной энергии при отклонение длины связи от равновесной. В классической теории силового поля термин растяжение связи представлен гармонической функцией.
Рентгеноструктурный анализ -рентгендифракционное исследование дает трехмерную атомную структуру веществ в кристаллическом состоянии. Эта структура отвечает минимальной энергией конформационных состояний или близкой к минимуму энергии. Атомные координаты, основанные на данных рентгеновской дифракции, часто используются для подготовки входных данных для квантово-химических расчетов. Они также могут служить как первичные данные для конформационного анализа. Анализ молекулярной упаковки дает также информацию о межмолекулярных контактах и межмолекулярных водородных связях.
РМХ (англ. - EHT) - расширенный метод Хюккеля - о дин из простейших полуэмпирических методов. Сформулирован Р. Хоффманном в начале шестидесятых годов. Не является методом самосогласованного поля. Расширенным методом Хюккеля получаюткачественное или полуколичественное описание молекулярных орбиталей и электронных свойств (например, заряды на атомах и распределение спиновой плотности). Удовлетворительно описывает распределение электростатического молекулярного потенциала (МЭСП). Не пригоден для оптимизации геометрии или молекулярно-динамических вычислений.
PM3 (параметрическая модель 3) - версия параметризации метода AM1; дает лучшие оценки теплот образования.
Свободная валентность – разность между максимально возможным полным порядком связи атома и его полным порядком связи в конкретном соединении. Отражает неполноту насыщения валентных возможностей данного атома.
Сгруппированные орбитали (СОГТ) – базисные функции, которые является линейной комбинацией гауссовых примитивов.
Секулярное уравнение – уравнение, возникающее при вариационном решении уравнения Шредингера путем минимизации выражения для энергии по варьируемым параметрам (коэффициентам разложения многоэлектронной волновой функции по частным волновым функциям φi):
| Hij - ESij| = 0.
Здесь Hij – матричный элемент оператора Гамильтона в базисе некоторых функций φi, Е – энергия состояния системы, Sij – элемент матрицы интегралов перекрывания функций φi.
Силовое поле - используется в молекулярной механике, представляет собой функцию потенциальной энергии молекулы от координат ядер атомов (силы, действующие на атомы, представляются в виде функций координат атомов). Определяется через потенциальные функции (представляющими собой, например, суммы парных потенциалов взаимодействия атомов), которые содержат некоторые параметры, численное значение которых выбирается оптимальным образом так, чтобы получить согласие рассчитанных и экспериментальных характеристик молекулы. В простейшем случае параметрами являются равновесные межъядерные расстояния (длины связей) и валентные углы, а также силовые постоянные, то есть коэффициенты жесткости упругих сил, связывающих пары атомов. При использовании в молекулярной механике основываются на допущении возможности переноса этих параметров из одной молекулы в другую, так что численные значения параметров, подобранные для некоторых простых молекул, используются далее при прогнозировании свойств других более сложных соединений.
Силовая постоянная - коэффициент пропорциональности между возвращающей силой и смещением x простого гармонического осциллятора; сила = - k x. Большие силовые постоянные отвечают жестко связанным системам (возвращающие силы велики даже при малых отклонениях от положения равновесия). Частота колебаний определяется не только силовой постоянной, но и массой системы, так как чем больше масса, тем меньше эффективна возвращающая сила. Силовая постоянная является мерой жесткости связей между атомами и определяет (наряду с массой атомов) колебательные частоты молекул.
Собственная функция – функция называется собственной функцией оператора, если в результате его действия на данную функцию получается та же функция, умноженная на число.
Спин-орбиталь – одноэлектронная функция ci (xi), в аргумент которой вводят спиновую переменную s (x i = r i | si). Пренебрегая малым по величине спин-орбитальным взаимодействием, каждую спин-орбиталь ci(x) можно представить в виде произведения пространственной орбитали ci(r) и спиновой функции h(s): ci(x) = ci(r)h(s).
Спин-орбитальное взаимодействие – взаимодействие между спиновым и орбитальным магнитным моментами электрона. Сила взаимодействия зависти от их взаимной ориентации, а энергия этого взаимодействия проявляется в спектрах как тонкая структура.
Спиновые корреляции – относительное расположение электронов коррелировано со взаимным направлением их спинов. Дополнительно к кулоновскому отталкиванию, электроны с параллельными спинами проявляют тенденцию избегать друг друга. В этом проявляется внутреннее свойство электронов, которое следует из принципа Паули. Такое поведение влияет на среднее отталкивание двух электронов и на обменную энергию. Вероятность того, что электроны с параллельными спинами находятся в одной и той же точке пространства, равна нулю. Область пространства, в которой находится электрон с определенным направлением спина и в которую электроны с таким же направлением спина стремятся не попадать, называется дыркой Ферми.
STO-n G - сокращение, используемое в расчетах аb initio. Указывает используемый базисный набор, в котором орбитали слейтеровского типа, аппроксимированы n гауссианами. STO-n G (где n = 3, 4) - минимальный базисный набор; используется редко.
Спиновая плотность – разность между плотностями электронов с противоположными спинами. Для систем с закрытой оболочкой спиновая плотность равна нулю в каждой точке. Спиновая плотность на ядре определяет спектр электронного парамагнитного резонанса.
Структурная область – область, соответствующая минимуму на поверхности потенциальной энергии, внутри которой сохраняется стабильная конфигурация ядер, соответствующие ей распределение электронной плотности и система химических связей.
Суммарный заряд системы – суммарный избыток заряда ядра над электронным зарядом (или наоборот).
Суперпозиционная ошибка базисных наборов (англ. BSSE - basis set superpositional error) - вычисляя энергию взаимодействия молекул вариационным методом, следует скорректировать ошибку, возникающую из-за использования более широкого набора базисных функций, относящихся ко всем молекулам комплекса, по сравнению с базисом, в котором рассчитывалась каждая молекула в отдельности. Игнорирование этого факта ведет к искусственному занижению энергии комплекса, т.е. к завышенной энергии взаимодействия. Чтобы исправить эту ошибку, энергию каждой отдельной молекулы рассчитывают, включая в базис функции, центрированные на атомных центрах соседних молекул. При этом полагают, что заряды ядер последних равны нулю и берут число электронов, равным таковому в отдельной рассматриваемой молекуле.
Супрамолекулярная химия – область химии, изучающая химическое, физическое и биологическое поведение молекулярных ансамблей, обусловленное невалентными межмолекулярными взаимодействиями.
Сферические гармоники – набор функций угловых координат θ и φ, удовлетворяющих дифференциальному уравнению Λ 2 Υ lm (θ, φ) = - l(l+1) Υ lm (θ, φ), где Λ2 – оператор Лежандра. Их можно выразить в виде полиномов от sinθ, cosθ, sinφ, cosφ или в виде полиномов от переменных x, y, z. Угловые зависимости сферических гармоник и граничные поверхности атомных орбиталей не вполне идентичны. Последние определяются как границы областей пространства, заключающих в себе определенные значения амплитуды или плотности, и зависимость этих характеристик от радиуса (а не только от углов) искажает форму сферических гармоник.
Т
Теорема Бриллюэна - матричные элементы электронного гамильтониана между основной Y0 и однократно-возбужденной Y1 конфигурациями равны нулю. Позволяет понизить число вычисляемых матричных элементов гамильтониана между конфигурациями Y0 и Yk. Это означает, что однократно возбужденные состояния молекул с заполненной оболочкой непосредственно не смешиваются с основным состоянием, что свидетельствует о стабильности основного состояния, вычисленного методом самосогласованного поля.
Теорема вириала – устанавливает соотношение между полной энергией многоэлектронной системы (Е) и ее кинетической (G) и потенциальной компонентами (V): -V = 2G и Е = - G. Справедлива как для полной системы, находящейся в равновесии, так и для частей, из которых она образуется (для молекул и атомов).
Теорема Гельмана-Фейнмана – для точной и хартри-фоковской волновой функции справедливо соотношение 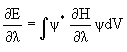 , где l - некоторый параметр, от которого зависит энергия. Применение соотношения этого соотношения к изучению сил, действующих в молекуле известно под названием электростатической теоремы:
, где l - некоторый параметр, от которого зависит энергия. Применение соотношения этого соотношения к изучению сил, действующих в молекуле известно под названием электростатической теоремы:
 =Za
=Za 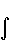 ρ(
ρ(  )
)  dV +
dV + 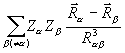 ( для ядра a молекулы), где ρ() – электронная плотность. Производная
( для ядра a молекулы), где ρ() – электронная плотность. Производная 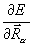 описывает силу, действующую на ядро в молекуле. Ее компонента по одной из осей координат равна
описывает силу, действующую на ядро в молекуле. Ее компонента по одной из осей координат равна
Fxα = – Zα 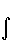 ρ( )
ρ( ) 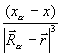 dV +
dV + 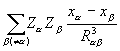 .
.
Сила, действующая на ядро а, есть сумма электростатического взаимодействия ядра а с электронной плотностью r и с другими ядрами. Пространство молекулы можно разделить на связывающую область, в которой электронная плотность создает электростатические силы, действующие на ядра по направлению друг к другу, и на антисвязывающую область, в которой силы, действующие на ядра, стремятся их раздвинуть. Такое разделение используется при анализе химической связи.
|
|
|
|
|
Дата добавления: 2015-06-25; Просмотров: 266; Нарушение авторских прав?; Мы поможем в написании вашей работы!