КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Методы систематического хода анализа катионов. 4 страница
|
|
|
|
ОСАЖДЕННАЯ И ГРАВИМЕТРИЧЕСКАЯ ФОРМЫ.
ТРЕБОВАНИЯ К НИМ.
В гравиметрическом методе осаждения существуют понятия осажденной
и гравиметрической форм вещества. Осажденной формой называют соединение, в виде которого определяемый компонент осаждается из раствора. Гравиметрической (весовой) формой называют соединение, которое взвешивают. Иначе ее можно определить как осажденную форму после соответствующей аналитической обработки осадка. Представим схемы гравиметрического определения ионов SO42-, Fe3+, Мg2+
S042- + Ва2+ ↔ BaS04↓ → BaS04 ↓
определяемый осадитель осажденная гравиметрическая
ион форма форма
Fe3+ + 3OH‾ ↔ Fe(OH)3↓ → Fe2O3↓
определяемый осадитель осажденная гравиметрическая
ион форма форма
Mg2+ + НРО4 2 -+ NH4∙H2O ↔ Mg NH4 P04↓ + H2O → Mg2 P2 O7 определ. осадитель осажденная форма гравиметрич. форма
ион
Из приведенных примеров видно, что не всегда гравиметрическая форма совпадает с осажденной формой вещества. Различны и требования, предъявляемые к ним.
Осажденная форма должна быть:
достаточно малорастворимой, чтобы обеспечить практически полное
выделение определяемого вещества из раствора. В случае осаждения
бинарных электролитов (AgCl; BaS04; СаС2О4 и т. п.) достигается
практически полное осаждение, так как произведение растворимости этих
осадков меньше, чем 10 - 8;
полученный осадок должен быть чистым и легко фильтрующимся (что определяет преимущества кристаллических осадков);
осажденная форма должна легко переходить в гравиметрическую форму.
После фильтрования и промывания осажденной формы ее высушивают или прокаливают до тех пор, пока масса осадка не станет постоянной, что подтверждает полноту превращения осажденной формы в гравиметрическую и указывает на полноту удаления летучих примесей. Осадки, полученные при осаждении определяемого компонента органическим реагентом (диацетилдиоксимом, 8-оксихинолином, α-нитрозо-β-нафтолом и т. д.), обычно высушивают. Осадки неорганических соединений, как правило, прокаливают
|
|
|
Основными требованиями к гравиметрической форме являются:
точное соответствие ее состава определенной химической формуле;
химическая устойчивость в достаточно широком интервале температур, отсутствие гигроскопичности;
как можно большая молекулярная масса с наименьшим содержанием
в ней определяемого компонента для уменьшения влияния погрешностей
при взвешивании на результат анализа.
ВЫЧИСЛЕНИЕ РЕЗУЛЬТАТОВ
В ГРАВИМЕТРИЧЕСКОМ МЕТОДЕ АНАЛИЗА
Гравиметрический анализ включает два экспериментальных измерения: определение массы навески mн анализируемого вещества и массы продукта известного состава, полученного из этой навески, то есть массы гравиметрической формы mгр.ф анализируемого вещества.
На основании этих данных несложно вычислить массовую процентную долю w, % определяемого компонента в навеске:
w, % = mгр.ф ∙ F ∙ 100 / mн,
где F - гравиметрический фактор (фактор пересчета, аналитический множитель) рассчитывают как отношение молекулярной массы определяемого компонента к молекулярной массе гравиметрической формы с учетом стехиометрических коэффициентов.
Значение гравиметрических факторов, рассчитанное с высокой точностью, приводится в справочной литературе.
Пример 1. Сколько граммов Fе2О3 можно получить из 1,63 г Fе3О4? Рассчитайте гравиметрический фактор.
Р е ш е н и е. Необходимо допустить, что Fе3О4 количественно превращается в Fе2О3 и для этого имеется достаточное количество кислорода:
2 Fе3О4 + [О] ↔ 3 Fе2О3
Из каждого моля Fе3О4 получается 3/2 моля Fе2О3. Таким образом, число молей Fе2О3 больше, чем число молей Fе3О4, в 3/2 раза, то есть:
|
|
|
nM(Fе2О3) = 3/2 nM(Fе3О4);
m(Fе2О3) / М(Fе2О3) = 3/2 m(Fе3О4) / М(Fе3О4)
где n - число молей определяемого компонента, из которого получается один моль гравиметрической формы; m - масса вещества, г; М - молярная масса вещества, г/моль.
Из формулы m(Fе2О3) = 3/2 (m(Fе3О4) ∙ М(Fе2О3)) / М(Fе3О4)
получаем
m(Fе2О3) = m(Fе3О4) ∙ 3М(Fе2О3) / 2М(Fе3О4)
и подставляем в нее численные значения:
m(Fе2О3) = 1,63 ∙(3 ∙ 159,7) / (2 ∙ 231,5) = 1,687 ≈ 1,69 г.
Гравиметрический фактор F равен:
F = 3М(Fе2О3) / 2М(Fе3О4) = 1,035.
Следовательно, в общем случае гравиметрический фактор определяют по формуле:
F = (а ∙ Мопред.в-во) / (b ∙ Мгр.ф),
где а и b - небольшие целые числа, на которые нужно умножить молекулярные массы, чтобы число молей в числителе и знаменателе было химически эквивалентно.
Однако не во всех случаях эти расчеты применимы. При косвенном определении железа в Fе2(SО4)3, которое заключается в осаждении и взвешивании BaSО4 (гравиметрическая форма), при расчете аналитического фактора в числителе и знаменателе формулы нет общего элемента. Здесь необходим другой способ выражения химической эквивалентности между этими величинами:
2 M(Fe3+) ≡≡ l М(Fе2(SО4)3) ≡≡ 3 M(SO42-) ≡≡ 3 M(BaSО4).
Гравиметрический фактор для массовой процентной доли железа будет выражаться:
F = 2M(Fe3+) / 3M(BaSО4).
Пример 2. Раствор препарата Nа3РО4 (mн = 0,7030 г) осадили в виде MgNН4РО4∙ 6Н2О. После фильтрования и промывания осадок прокалили при 1000 ˚С. Масса полученного осадка Mg2P2О7 составила 0.4320 г. Рассчитайте массовую процентную долю фосфора в навеске
Р е ш е н и е.
mгр.ф (Mg2P2О7) = 0,4320 г;
F = 2М(Р) / М(Mg2P2О7) = 0,2782; mн = 0,7030 г;
W,% = mгр.ф ∙ F ∙ 100 / mн
w, %(Р) = 0,4320 ∙ 0,2782 ∙ 100 / 0,7030 = 17,10 %.
Пример 3. При прокаливании загрязненного препарата натрия оксалата mн = 1,3906 г получили остаток массой mгр.ф = 1,1436 г. Определите степень чистоты образца. t
Na2C2О4 → Nа2СО3 + СО↑
Ре ш е н и е. Следует допустить, что разница между исходной и конечной массами соответствует потере углерода оксида при прокаливании. Анализ основан на измерении этой величины:
n(СО) = n(Na2C2O4),
следовательно,
w, %(Na2C2O4) = (mн - mгр.ф) ∙ F ∙ 100 / mн;
F = M(Na2C2O4) / M(CO) = 4,784;
w, %(Na2C2O4) = (1,3906 – 1,1436) ∙ 4,784 ∙ 100 / 1,3906 = 84,97 %.
ВЫБОР МАССЫ НАВЕСКИ В ГРАВИМЕТРИИ
|
|
|
Как известно, точность анализа зависит как от массы навески, так и от массы гравиметрической формы, получаемой из нее. Если навеска будет взята с большой точностью, а полученная из нее гравиметрическая форма будет малой величиной, измеренной с большой погрешностью, то весь анализ будет выполнен с ошибкой, допущенной при взвешивании гравиметрической формы. Поэтому должна быть взята такая навеска, чтобы при ее взвешивании и при взвешивании полученной из нее гравиметрической формы ошибка не превышала ± 0,2 %. Для этого необходимо определить минимальную массу, которую еще можно взвесить с точностью ± 0,2 % на аналитических весах с абсолютной ошибкой взвешивания ± 0,0001 г, а минимальная ошибка, учитывая возможный разброс (±), в этом случае будет равной 2 ∙ (±0,000 1) = ±0,0002 г.
100 г - ± 0,2 г
х - ± 0,0002 г
х = 0,1 г
Следовательно, такой минимальной массой mmin является 0,1 г. При величине, меньшей чем 0,1 г, ошибка превысит 0,2 %. При расчете массы навески в гравиметрическом анализе масса гравиметрической формы компонента приравнивается к минимальной массе вещества:
mгр.ф = mmin, mн = mmin ∙ F ∙ 100 / w, %.
Если величина массы навески, рассчитанная по указанной формуле, окажется менее 0,1 г, то навеску следует увеличить до 0,1 г. Чаще всего массу исходной навески указывают в методике анализа или же для объемных аморфных осадков массу навески берут около 0,1, а для кристаллических от 0,1 до 0,5 г.
Расчет количества осадителя проводят с учетом возможного содержания определяемого компонента в анализируемой пробе. Для полноты выделения осадка применяют умеренный избыток осадителя. Если осадитель летуч (например, раствор хлороводородной кислоты), берут двух-, трехкратный избыток, который впоследствии удаляют при нагревании осадка. Если осадитель нелетуч (растворы бария хлорида, аммония оксалата, серебра нитрата и т. п.), достаточно его полуторакратного избытка.
АНАЛИТИЧЕСКИЕ ВЕСЫ. ПРАВИЛА ОБРАЩЕНИЯ С НИМИ
Аналитические весы - это точный физический прибор, пользование которым допускается при строгом соблюдении правил, обеспечивающих необходимую воспроизводимость и точность взвешивания.
|
|
|
Правила обращения с аналитическими весами включают следующие основные требования:
1. Весы должны быть установлены на жестко закрепленной поверхности,
зaщищающей их от различных потрясений, и в специально оборудованном помещении - весовой комнате.
2. Недопустимы резкие колебания температуры, действие прямых солнечных лучей, а также воздействие на аналитические весы химических веществ.
3. Предельно допустимая нагрузка аналитических весов должна быть не более 200 г.
4. При взвешивании предметов на аналитических весах необходимо, чтобы они имели температуру весовой комнаты.
5. Взвешиваемое вещество помещают на левую чашку весов в специальной таре (бюксы, тигли, часовое стекло). Гири аналитического разновеса помещают на правую чашку весов.
6. Взвешиваемые предметы и гири вносят через боковые дверцы весов (шторки). Взвешивание производят только при закрытых дверцах весов.
7. Гири аналитического разновеса берут только специально предназначенным пинцетом. Все операции со сменой разновеса производят при полном арретировании весов.
8. До и после каждого взвешивания необходимо проверять нулевую точку весов.
9. Во избежание перекоса чашек весов гири и взвешиваемые предметы помещают в центр чашек.
10. Запись результатов взвешивания проводят по пустым гнездам аналитического разновеса и по данным барабанов с десятыми и сотыми долями грамма. Третий и четвертый знаки после запятой снимают со светящегося табло.
11. По окончании взвешивания необходимо убедиться, что весы арретированы, полностью разгружены и дверцы футляра плотно закрыты.
12. Для уменьшения ошибки взвешивания необходимо пользоваться аналитическим разновесом, предназначенным для строго определенных аналитических весов.
Следует отметить, что даже при соблюдении всех упомянутых правил
могут возникать ошибки взвешивания, зависящие от различных причин:
вызванные неравноплечестью коромысла весов;
за счет изменения массы тела в процессе взвешивания;
за счет взвешивания в воздухе, а не в вакууме;
вызванные несоответствием массы гирь (разновесов) их номинальной
массе.
ПРИМЕНЕНИЕ ГРАВИМЕТРИЧЕСКОГО МЕТОДА АНАЛИЗА
Использование неорганических осадителей позволяет получать в виде гравиметрической формы либо соли, либо оксиды определяемых веществ. Неорганические реагенты не отличаются специфичностью, но в анализе наиболее часто используют: NH4ОH (Fе2О3, SnО2); H2S (CuS, ZnS или ZnSО4, As2S3 или As2S5, Вi2S3); (NH4)2S (HgS); NH4H2PО4 (Mg2P2О7, Al3PО4, Мn2Р2О7); H2SО4 (PbSО4, BaSО4, SrSО4); Н2С2О4 (СаО); НСl (AgCl, Hg2Cl2, Na в виде NaCl из бутанола); AgNО3 (AgCl, AgBr, AgI); BaCl2 (BaSO4) и пр.
Иногда в основу гравиметрических определений положено восстановление определяемого компонента до элемента, который служит гравиметрической формой.
Для гравиметрического определения неорганических веществ предложен ряд органических реагентов, которые, как правило, обладают большей селективностью. Известны два класса органических реагентов. Первые образуют малорастворимые комплексные (координационные) соединения и содержат не менее двух функциональных групп, имеющих пару неподеленных электронов. Еще их называют хелатообразующими реагентами, например 8-оксихинолин осаждает более двадцати катионов:
N
OH
Растворимость оксихинолятов металлов изменяется в широких пределах в зависимости от природы катиона, значения рН среды.
В 1885 году бьл предложен l-нитрозо-2-нафтол - один из первых селективных органических реагентов, который широко используют для определения кобальта в присутствии никеля, а также для определения ионов висмута(3), хрома (III), ртути (II), олова (IV) и т. п.:
NO
OH
Диацетилдиоксим (диметилглиоксим) отличается высокой селективностью, и его широко используют для гравиметрического определения малых концентраций никеля:
CH3 ─ C ─ C ─ CH3
│ │
OH - N N - OH
ПОГРЕШНОСТИ ГРАВИМЕТРИИ
Гравиметрический метод анализа дает наиболее правильный результат, и, несмотря на длительность и трудоемкость, его очень часто применяют как проверочный метод в арбитражных анализах. Систематические методические ошибки в гравиметрии могут быть учтены и уменьшены в ходе выполнения соответствующих операций (табл. 1.2).
Методические погрешности гравиметрии
| Гравиметри-ческая операция | Абсолютная погрешность | |
| положительная (завышенный результат) | отрицательная (заниженный результат) | |
| Выбор осадителя: а) природа осадителя б) количество осадителя | Нелетучий, неспецифический осадитель Небольшой избыток осадителя, соосаждение посторонних ионов | Высокая растворимость осаждаемой формы, коллоидообразование Недостаток осадителя. Слишком большой избыток осадителя, повышение растворимости осадка в результате комплексообразования или солевого эффекта |
| Осаждение | Соосаждение посторонних ионов | Недостаточное время созревания (кристаллические осадки). Коллоидообразование (аморфные осадки) |
| Фильтрование | ________ | Неправильный выбор фильтра – прохождение частиц осадка через фильтр |
| Промывание | Промывание нелетучей промывной жидкостью | Избыток промывной жидкости: пептизация аморфного осадка; гидролиз кристаллического осадка. Потери в результате растворимости |
| Получение гравиметри-ческой формы | Температура прокаливания: получение соединения другого состава, гигроскопичность, поглощение СО2 из воздуха | Превышение температуры высушивания для осадков органической природы. Превышение температуры прокаливания (получения соединения другого химического состава) |
Таблица 1.2
Правильность метода объясняется малой систематической ошибкой измерений, связанной с точностью взвешивания на аналитических весах:
Sx / x = √(Sa / a)2 + 1/n(Sm / m)2,
где Sa – точность взвешивания на аналитических весах (0,0002 г для весов АДВ – 200; 0,00005 г для полумикровесов и т.д.); а – навеска анализи-руемого вещества, г; т - масса гравиметрической формы, г; п - количество прокаливаний или высушиваний для получения постоянной массы.
Анализ приведенных данных показывает, что выявить вид ошибки можно при рассмотрении методики определения с учетом механизма образования осадка, свойств веществ, используемых и получающихся в ходе анализа.
В настоящее время значение гравиметрических методов анализа несколько уменьшилось, однако не следует забывать, что, имея достоинства и недостатки, гравиметрический анализ является оптимальным для решения достаточно большого количества аналитических задач.
Аналитическая химия - лекция №7
КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ. СУЩНОСТЬ МЕТОДА И ЕГО ВОЗМОЖНОСТИ. ИНТЕРВАЛ ПЕРЕХОДА ОКРАСКИ КИСЛОТНО-ОСНОВНЫХ ИНДИКАТОРОВ. КРИВЫЕ ТИТРОВАНИЯ.
Метод кислотно-основного титрования основан на реакциях взаимодействия между кислотами и основаниями, то есть на реакции нейтрализации:
Н + + ОН - ↔ Н2О
Рабочими растворами метода являются растворы сильных кислот (HCl, H2S, НNОз и др.) или сильных оснований (NaOH, КОН, Ва(ОН)2 и др.). В зависимости от титранта метод кислотно-основного титрования подразделяют на ацидиметрию, если титрантом является раствор кислоты, и алкалиметрию, если титрантом является раствор основания.
Рабочие растворы в основном готовят как вторичные стандартные растворы, поскольку исходные для их приготовления вещества не являются стандaртными, а затем их стандартизуют по стандартным веществам или стандартным растворам. Например: растворы кислот можно стандартизовать по стандартным веществам - натрия тетраборату Na2B4О7 ∙10Н2О, натрия карбонату Nа2СО3 ∙10Н2О или по стандартным растворам NaOH, КОН; а растворы оснований - по щавелевой кислоте Н2С2О4 ∙ 2Н2О, янтарной кислоте Н2С4Н4О4 или по стандартным растворам HCl, H2SO4, НNО3.
Точка эквивалентности и конечная точка титрования. Согласно правилу эквивалентности титрование необходимо продолжать до тех пор, пока количество прибавленного реагента не станет эквивалентным содержанию определяемого вещества. Наступающий в процессе титрования момент, когда количecтвo стандартного раствора реагента (титранта) становится теоретически строго эквивалентным количеству определяемого вещества согласно определенному уравнению химической реакции, называют точкой эквивалентности.
Точку эквивалентности устанавливают различными способами, например по изменению окраски индикатора, приба-вляемого в титруемый раствор. Момент, при котором происходит наблюдаемое изменение цвета индикатора, называют конечной точкой титрования. Очень часто конечная точка титрования не совсем совпадает с точкой эквивалентности. Как правило, они отличаются друг от друга не более чем на 0,02-0,04 мл (1-2 капли) титранта. Это то количество титранта, которое необходимо для взаимодейcтвия с индикатором.
ИНДИКАТОРЫ В МЕТОДЕ КИСЛОТНО-ОСНОВНОГО ТИТРОВАНИЯ
В методах кислотно-основного титрования для определения конечной точки титрования используют кислотно-основные индикаторы. Кислотно-основные индикаторы - это органические вещества, способные видимо и обратимо изменять свою окраску в растворе при изменении рН среды. Существуют различные теории индикаторов, каждая из которых по-своему объясняет поведение кислотно-основных индикаторов в кислых и щелочных средах.
Ионная теория индикаторов. В связи с тем, что кислотно-основные индикаторы представляют собой слабые кислоты или слабые основания, любой индикатор диссоциирует в растворе согласно уравнению:
HInd ↔ Н+ + Ind-
бесцветный малиновый
Окраска раствора, в котором индикатор находится в молекулярной форме (HInd), отличается от окраски раствора, в котором индикатор находится в ионной форме (Ind -). Так, моле-кулы фенолфталеина HInd бесцветны, а его анионы Ind - окрашены в малиновый цвет. Достаточно к раствору, содержащему фенолфталеин, прибавить 1-2 капли щелочи, как введенные ОН--ионы станут связывать катионы Н+ с образованием слабого электролита - молекул воды. При этом равновесие диссоциации индикатора сместится вправо, и накопление анионов Ind- вызовет окрашивание раствора в малиновый цвет.
Переход одной окраски, присущей молекулярной форме кислотно-основного индикатора, в другую, свойственную его ионной форме, происходит под влиянием Н+ или ОН--ионов, то есть зависит от рН раствора.
Хромофорная теория индикаторов. Поведение индикаторов, объясняемое ионной теорией индикаторов, дополняется хромо-форной теорией индикаторов, согласно которой изменение окраски индикаторов связано с изменением структуры их молекул, внутримолекулярной перегруппировкой, вызываемой действием Н+ или ОН--ионов. По хромофорной теории в процессе изменения рН раствора меняется строение молекул кислотно-основных индикаторов. Это явление обусловливается бензоидно-хиноидной таутомерией. При изменении рН среды раствора или при диссоциации хромофоры могут перегруппировываться. Перемена окраски у индикаторов является результатом изменений в их внутреннем строении. У одноцветных индикаторов окраска изменяется в связи с появлением или исчезновением хромофоров. У двухцветных индикаторов эти изменения обусловлены превращением одних хромофоров в другие.
Типичным одноцветным индикатором является фенол-фталеин. При рН < 8 его молекулы не содержат хиноидной груп-пировки и поэтому бесцветны. Однако при добавлении раствора щелочи к раствору фенолфталеина (рН = 8) происходит перегруппировка атомов в молекуле с образованием хиноидной группировки, которая обусловливает появление малиновой окраски раствора:
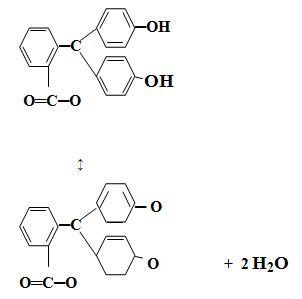
Дальнейшее увеличение рН до 13-14 вызывает другую пере-группировку, в результате чего получается трехзамещенная соль, лишённая хиноидной группировки и поэтому бесцветная:
Вследствие этого фенолфталеин обесцвечивается при действии большого избытка щелочи, например, натрия гидроксида. Типичным двухцветным индикатором является метиловый оранжевый:
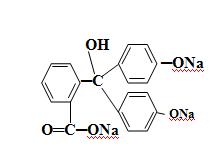
(CH3)2N N═N SO3Na
При рН = 3,2...4,3 он оранжевый, при рН ≤ 3,1 приобретает красную, а при рН ≥ 4,4 - желтую окраску. Это объясняют присоединением ионов водорода кислоты к атому азота азогруппы индикатора, вследствие чего происходит смещение электронов по всей системе, сопровождающееся образованием хиноидной структуры, которая обусловливает появление красной окраски раствора. Таким образом, при действии кислот наблюдают переход желтой окраски индикатора в красную, а при действии щелочей - обратное превращение:
(CH3)2N ═ ═N―N
ОН ↔
[(CH3)2N N═N SO3 ] + Н2 О
Цветность органических соединений, согласно хромофорной теории, обусловливается не только хиноидной структурой молекул, но и присутствием в них других хромофорных группировок (-N=N-, -N02, -NO, =С=С=, =С=О). При введении в молекулы органических веществ, содержащих хромофорные группы, ряда других групп - ауксохромов (-ОН, -Nh4, -NHR, -NHR) происходит углубление цвета окрашенного вещества.
Ионно-хромофорная теория индикаторов. Согласно дополняющим друг друга ионной и хромофорной теориям, в раст-ворах кислотно-основных индикаторов одновременно сосущест-вуют равновесия, обусловливаемые диссоциацией молекул, и равновесия, связанные с внутримолекулярными перегруппировками (ионно-хромофорная теория). Для кислотно-основных индика-торов наиболее характерными факторами, вызывающими измене-ние окраски, являются изменение соотношения количеств молеку-лярной и ионной форм индикатора, происходящее под влиянием кислот и щелочей, и появление или исчезновение хромофорных групп или же превращение одних хромофорных групп в другие.
Способность молекул различных индикаторов диссоциировать в нейтральной среде характеризуют константами диссоциации. Например, у метилового оранжевого Кa≈ 10 -4, у лакмуса Кa ≈ 10 -8, а у фенолфталеина Кa ≈ 10-9. Следовательно, фенолфталеин является наиболее слабой органической кислотой из этих индикаторов.
Известно, что прибавление к любому раствору любой кислоты или щелочи влечет за собой изменение концентрации ионов Н+ в нем, а следовательно, и величины рН. Перемена окраски у индика-торов также связана с изменением рН раствора. Однако каждый индикатор изменяет окраску только в определенном, характерном для него интервале значений рН. Объясняется это тем, что окраска индикатора зависит от соотношения концентраций его диссоци-ированной и недиссоциированной форм, то есть от отношения:
KHInd=[H+][Ind-] / [HInd]
[Ind-] / [HInd] = KHInd / [H+] или [HInd] / [Ind-] = [H+] / KHInd.
Когда KHInd = [Н+], то [Ind-] / [HInd] = 1.
Если КHInd / [Н+] > 1, то в растворе превалирует диссоцииро-ванная форма индикатора, а если КHInd / [Н+] < 1, то превалирует недиссоциированная форма.
При одной и той же концентрации ионов водорода отношение КHInd / [Н+] будет тем больше, чем больше КHInd.
Для фенолфталеина КHInd = [Н+] [Ind-] / [HInd] ≈ 10-9.
При рН = 7 [Н+] = 10 -7, а [HInd] / [Ind-] = 10-7 / 10-9, то есть при рН = 7 на каждые 100 бесцветных молекул фенолфталеина приходится лишь 1 окрашенный ион, следовательно, раствор - бесцветный. Если к раствору фенолфталеина прибавить щелочь и довести рН раствора до 8, то соотношение [HInd] / [Ind -] = 10 -8 /10-9 (уменьшится в 10 раз), и раствор станет бледно розовым. А при рН=9 соотношение [HInd] / [Ind-] = 10-9 / 10-9 = 1, то есть в растворе присутствуют равные количества бесцветных молекул индикатора и окрашенных в красный цвет ионов и раствор приобретает розовую окраску.
Таким образом, переходная окраска индикатора появляется при рН среды, равном рКHInd, но так как изменение цвета индикатора происходит постепенно, цвет недиссоциированных молекул индикатора начинает маскироваться цветом ионов задолго до достижения соотношения [HInd] / [Ind-] = 1.
Следовательно, цвет водного раствора индикатора определяется соотношением концентрации его молекулярной и ионной форм, отличающихся различной окраской, и зависит от [Н+]. Величину рН, до которой титруют раствор с данным индикатором, называют показателем титрования этого индикатора рТ.
Важнейшие индикаторы имеют следующие области перехода и показатели титрования:
Показатель титрования рТ Область перехода рН
Метиловый оранжевый…4,0…………… 3,1 - 4,4
Метиловый красный….. 5,5…………… 4,4 - 6,2
Лакмус……………… ……7,0…………… 5,0 - 8,0
Фенолфталеин……………9,0………….. 8,0 - 10,0
КРИВЫЕ ТИТРОВАНИЯ. ВЫБОР ИНДИКАТОРА
Кривая кислотно-основного титрования - это графическое изображение изменения рН раствора в ходе титрования.
Титрование сильной кислоты сильным основанием.
Допустим, что для титрования взяли 20 см3 раствора 0,1 моль/дм3 HCl, а в качестве титранта использовали раствор 0,1 моль/дм3 NaOH. Поскольку каждая молекула НСl дает при диссоциации один ион Н+, общая концентрация водородных ионов в 1 дм3 исходной 0,1 моль/дм3 кислоты составляет 0,1 (или 10 -1) моль-ион. Следовательно, рН этого раствора равен 1.
Когда 90 % соляной кислоты будет оттитровано, ионов Н+ останется 10 % от первоначального количества, то есть 0,01 (или 10-2) моль-ион в 1 дм3, а рН раствора станет равен 2. При нейтрализации 99,0 % соляной кислоты рН = 3; при нейтрализации 99,9 % кислоты рН = 4 и т. д. В момент полной нейтрализации соляной кислоты титруемый раствор содержит только натрия хлорид и имеет рН = 7. Прибавление избытка натрия гидроксида ведет к увеличению рН раствора, как это показано в табл. 3.1.
Результаты этих вычислений изображают графически. На оси абс-цисс откладывают избыток кислоты или щелочи в разные моменты титрования, а на оси ординат - соответствующие значения рН раствора. Получающийся график называют кривой титрования.
Ход этой кривой свидетельствует, что в конце титрования сильной кислоты сильным основанием происходит резкий скачок в измене-нии рН раствора. К моменту нейтрализации 99,9 % кислоты рН постепенно растет от 1 до 4, то есть всего на три единицы, а при переходе от 0,1 % остатка НСl к 0,1 % избытку NaOH рН раствора резко увеличивается с 4 до 10. Это означает, что добавление одной капли щелочи в конце титрования понижает концентрацию ионов Н+ с 10 -4 до 10 -10 моль в литре или в миллион раз.
Изменение рН раствора при титровании сильной кислоты сильным основанием
| Прибавлено NaOH | Осталось HCl | [H+], моль/дм3 | рН | |||||||
| % | мл | % | моль/дм3 | |||||||
| 0,0 90,0 99,0 99,9 | 0,0 18,00 19,80 19,98 | 100,0 10,0 1,0 0,1 | 0,1 0,01 0,001 0,0001 | 10 -1 10 -2 10 -3 10 -4 | ||||||
| 100,0 | 20,00 | 0,0 | 10 -7 | 10-7 | ||||||
| Избыток NaOH | ||||||||||
| 100,1 101,0 110,0 200,0 | 20,02 20,20 22,00 40,00 | 0,1 1,0 10,0 100,0 | 0,0001 0,001 0,01 0,1 | 10 -10 10 -11 10 -12 10 -13 | ||||||
В результате резкого изменения рН раствора от последней капли раствора основания происходит и резкое изменение окраски индикатора. При отсутствии скачка рН на кривой титрования окраска индикатора изменялась бы постепенно и определить точку эквивалентности было бы невозможно.
Титрование слабой кислоты сильным основанием
Кривую титрования слабой кислоты сильным основанием рассчитывают несколько иначе, так как при этом концентрацию ионов Н+ уже нельзя приравнивать к общей концентрации кислоты. Ее приходится вычислять с учетом константы диссоциации кислоты. Не вдаваясь в подробности вычислений, приведем кривую титрования раствора 0,1 моль/дм3 уксусной кислоты раствором 0,1 моль/дм3 NaOH (рис.3.2). Интервал скачка рН на ней значительно уже, чем в первом случае. Он простирается от рН = 7,8 (остатка кислоты в 0,1 %) до рН = 10 (∆pH = 2,2 избытка щелочи в 0,1 %). Слабая уксусная кислота посылает в раствор гораздо меньше ионов Н+, чем хлороводородная. Поэтому перед началом титрования рН раствора 0,1 моль/дм3 уксусной кислоты равен 3, а не 1, как в случае с хлороводородной кислотой. В ходе титрования рН раствора уксусной кислоты все время остается выше, чем при тех же концентрациях хлороводородной кислоты. Поэтому и скачок на кривой начинается с более высокого значения рН. Заканчивается он, как и впервом случае, при рН = 10, так как титрование производят тем же раствором 0,1 моль/дм3 NaOH.
|
|
|
|
Дата добавления: 2014-01-04; Просмотров: 389; Нарушение авторских прав?; Мы поможем в написании вашей работы!