КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Статистическая обработка результатов химического эксперимента
|
|
|
|
Аналитическая химия - лекция №15
Метрологические характеристики методов и результатов, которые получают при статистической обработке экспериментальных данных, позволяют проводить оценку и сравнение как экспериментальных методик, так и анализируемых объектов, и на этом основании решать прикладные задачи, связанные с определением статистической достоверности результатов исследований.
Ошибки в количественном анализе
Любое измерение имеет определенную ошибку, связанную с точностью измерительной аппаратуры, особенностями метода и случайными причинами. Во время анализа возникают ошибки при выполнении отдельных операций (взятии навески, растворении и т. д.).
Ошибки в количественном анализе делятся на систематические, случайные и промахи.
Систематические ошибки вызывают однотипные (одного знака) отклонения от истинного значения. Они зависят от особенностей данного метода анализа (методические ошибки), неточности измерений (инструментальные ошибки), недостаточной чистоты реагентов (реактивная ошибка), индивидуальных особенностей самого аналитика. Их можно выявить, уменьшить или внести поправки.
Случайные погрешности не имеют определенного знака, в их появлении отсутствуют любые закономерности. Их нельзя устранить введением каких-либо поправок, но они могут быть значительно уменьшены при повышении тщательности работы и увеличении числа параллельных определений.
Промахи — это грубые погрешности, возникающие при неверных измерениях, расчетах, неправильных записях и т. д. При обработке экспериментальных данных результаты с грубыми ошибками должны быть выявлены и отброшены.
По способу выражения погрешности количественных определений делят на абсолютные и относительные.
|
|
|
Абсолютной ошибкой (ΔХi) называют разность между полученным результатом (Xi) и истинным значением величины (μ), которую определяют как ΔХi = Xi - μ. При этом в большинстве случаев среднее выборки (X) является лучшей оценкой истинного значения измеряемой величины (μ):
Относительной ошибкой называется отношение абсолютной ошибки к истинному значению определяемой величины, выраженное в процентах:
n
X = ∑ Xi / n
Правильность, воспроизводимость и точность анализа
Правильность анализа показывает близость результата или среднего арифметического нескольких результатов X к истинному значению. Обычно ее обозначают погрешностью ДХ. Правильность анализа указывает на его качество, то есть практическое отсутствие систематической ошибки.
Воспроизводимость анализа определяют близостью параллельно полученных результатов и обозначают величиной отклонения полученных результатов от их среднеарифметического значения. Воспроизводимость анализа зависит от случайных ошибок.
Точность анализа отображает приближение к нулю ошибок всех видов.
Основные статистические характеристики, их вычисление. Проверка однородности выборки
Выборкой называют совокупность статистически эквивалентных результатов. Например, ряд результатов, полученных при параллельных определениях любого вещества в пробе.
Результаты, полученные при статистической обработке выборки, будут достоверны в том случае, если выборка однородна, то есть в полученных результатах анализа отсутствуют грубые ошибки.
Оценку однородности выборки удобно проводить по ^-критерию, если число опытов не превышает 10. Для этого все полученные результаты размещают в порядке возрастания: Xv X2,..., X.
Разность между самым большим (Хп) и самым малым (Х{) значениями называют размахом варьирования R = \X]— Xn\. Затем рассчитывают контрольные критерии Q для первого и последнего ре зул ьтато в в ы бор к и:
|
|
|
| Цифровые значения | контрольного критерия | Q (Р, и) | |||
| п | Q | ||||
| Р = 0,90 | Р = 0,95 | Р- 0,99 | |||
| 0,89 | 0,94 | 0,99 | |||
| 0,68 | 0,77 | 0,89 | |||
| 0,56 | 0,64 | 0,76 | |||
| 0,48 | 0,56 | 0,70 | |||
| 0,43 | 0,51 | 0,64 | |||
| 0,40 | 0,48 | 0,58 |
Значения Хх или Хя, для которых Q больше табличного значения Q{P, n), отбрасываются. Для выборки уменьшенного объема вновь по выше приведенным формулам выполняют цикл вычислений и проводят проверку однородности до получения однородной выборки сокращенного объема.
Основные статистические характеристики
В теории ошибок доказывается, что при выполнении нормального закона распределения случайных величин (закон Гаусса) среднее арифметическое всех результатов анализа (X) является лучшей оценкой истинного значения результата анализа µ:
п
X =(Х1 + Х2 + … +Хп) / п = (∑ Хi) / п.
i=1
Это среднее значение приблизительно равно истинному значению результатов анализа: X ≈ µ.
Рассеяние результатов анализа относительно среднего значения принято характеризовать д и с и е р с и е й S2:
п n
S 2 = ∑ (Хi – X)2 / f = (∑ Хi2 – nX2) / f
i=1 i=1
где f — число степеней свободы; f= n — 1.
Затем вычисляют среднее квадратичное отклонение S, которое рассматривают как оценку случайной ошибки, характерной для данной выборки:
n n
S = √ S2 = √(∑ (Хi – nX)2 / f = √ ∑ Хi2 – nX2 / f.
i =1 i=1
Стандартное отклонение среднего результата S% рассчитывают по уравнению:
S X = S / √ n.
Оценка доверительного интервала результатов анализа
Результаты анализа характеризуются доверительным интервалом среднего
значения ∆Х, которые рассчитывают по формуле:
∆X = t(P, f)∙ S X,
где t(P, f) — критерий Стьюдента (табличное значение), при числе степеней свободы f и доверительной вероятности Р = 0,95, принятой в аналитических расчетах (табл. 2.2).
Доверительный интервал ограничивает область, внутри которой при отсутствии систематических погрешностей находится истинное значение результата анализа с заданной доверительной вероятностью Р:
|
|
|
(Х - ∆Х) ≤ µ ≤ (Х + ∆Х).
Относительную ошибку среднего результата А (в процентах) рассчитывают по формуле:
A = ∆ X / X ∙ 100 %.
Пример 3. Рассчитать основные метрологические характеристики определения NaHCO3 методом кислотно-основного титрования:
ω, % = 24,11; 24,15; 24,18; 24,20.
Проверяем выборку на однородность. Для этого рассчитываем размах варьирования R:
R = | 24,11 – 24,20 | = 0,09,
Q1 = | Х1 – Х2 | / R = | 24,11 – 24,15 | / 0,09 = 0,44,
Q4 = | Х4 – Х3 | / R = | 24,20 – 24,18| / 0,09 = 0,22
при п = 4, Р = 0,95, Qтабл = 0,85 (данные табл. 2.1).
Q1 < Qтабл, Q4 < Qтабл - выборка однородна.
Вычисляем основные метрологические характеристики.
Среднее арифметическое результата:
_ п
Х = ∑ Хi = 24,11 + 24,15 + 24,18 + 24,20 / 4 = 24,16.
i = 1
Среднее квадратичное отклонение:
n
____________________________
S = √ ∑ Хi2 – пХ2 / f = √ 2334,82 – 4 · 24,162 / 3 = 2,83 · 10-2.
i = 1
Стандартное отклонение среднего результата:
__
SХ = S / √ п = 2,83 · 10-2 / 2 = 1,42 · 10-2.
Доверительный интервал среднего значения:
∆Х = t (P, f) · SX = 3,18 · 1,42 · 10-2 = 4,5 · 10-2 ≈ 0,05,
так как при Р = 0,95 и f = п-1 = 3, t (P, f) = 3,18 (табл. 2.2).
Относительная ошибка среднего результата:
А = (∆Х / Х) · 100 % = (4,5 · 10-2 / 24,16) · 100 % = 0,19 %.
Результат анализа записывают в виде:
ω NaHCO3, % = 24,1 (6) ± 0,05.
Необходимо заметить, что статистической обработке подлежат не результаты отдельных измерений, а конечные результаты анализа.
Аналитическая химия - лекция №16
Хроматографические методы анализа. Классификация методов, применение их в анализе.
Хроматография - наиболее часто используемый аналитический метод. Новейшими хроматографическими методами можно проанализировать газообразные, жидкие и твердые вещества с различной молекулярной массой. Это могут быть изотопы водорода, ионы металлов, полимеры, белки, нефть и др. С помощью хроматографии получена обширная информация о строении и свойствах многих классов органических соединений. Применение хроматографических методов для разделения белков оказало огромное влияние на развитие современной биохимии. Хроматографию с успехом применяют в исследовательских и клинических целях в самых разных областях биологии и медицины, в фармацевтике и криминалистике: для терапевтического мониторинга в связи с ростом нелегального употребления наркотиков, идентификации антибиотиков и отнесения их к той или иной группе антибактериальных препаратов, для анализа отдельных наиболее важных классов пестицидов. Такие достоинства, как универсальность, экспрессность и чувствительность делают хроматографию важнейшим аналитическим методом.
|
|
|
Хроматографический метод анализа разработан русским ботаником М.С. Цветом в 1903 г. В первых же работах с помощью этого метода М.СЦвет установил, что считавшийся однородным зеленый пигмент растений хлорофилл на самом деле состоит из нескольких веществ. При пропускании экстракта зеленого листа через колонку, заполненную порошком мела, и промывании петролейным эфиром он получил несколько окрашенных зон, что, несомненно, говорило о наличии в экстракте нескольких веществ. Впоследствии это было подтверждено другими исследователями. Этот метод он назвал хроматографией (от греч. "хроматос"- цвет), хотя сам же указал на возможность разделения и бесцветных веществ.
Компоненты анализируемой смеси (сорбаты) вместе с подвижной фазой передвигаются вдоль стационарной фазы. Ее обычно помещают в стеклянную или металлическую трубку, называемую колонкой. В зависимости от силы взаимодействия с поверхностью сорбента (за счет адсорбции или по какому-либо другому механизму) компоненты будут перемещаться вдоль колонки с разной скоростью. Одни компоненты останутся в верхнем слое сорбента, другие, в меньшей степени взаимодействующие с сорбентом, окажутся в нижней части колонки, а некоторые и вовсе покинут колонку вместе с подвижной фазой. Таким образом, происходит быстрое разделение сложных смесей компонентов. При перемещении вдоль колонки подвижная фаза встречает на своем пути все новые и новые слои сорбента, что обеспечивает многократность актов сорбции - десорбции разделяемых компонентов. Этим обусловлена значительно большая эффективность хроматографического разделения по сравнению со статическими методами сорбции и экстракции.
Хроматография- это физико-химический метод разделения и определения веществ, основанный на распределении компонентов между двумя фазами, неподвижной и подвижной. Неподвижной (стационарной) фазой служит твердое пористое вещество или пленка высококипящей органической жидкости, нанесенная на твердое вещество. Подвижная фаза представляет собой жидкость или газ, протекающий через неподвижную фазу.
Хроматография — гибридный метод анализа, в котором хроматографический процесс является частью общей аналитической системы, сочетающей разделение и измерение. Метод позволяет не только разделять многокомпонентную смесь, но идентифицировать и определять ее количественный состав.
Классификация методов хроматографии
Различные методы хроматографии можно классифицировать по агрегатному состоянию фаз, способу их относительного перемещения, аппаратурному оформлению процесса и т. д.
По агрегатному состоянию фаз хроматографические методы обычно классифицируют следующим образом (табл.1):
Таблица 1
| Неподвижная фаза | Подвижная фаза | |
| Газообразная | Жидкая | |
| Твердая | Газо-адсорбционная хроматография | Жидкостно-адсорбционная колоночная Ионообменная Осадочная |
| Жидкая | Распределительная газо- жидкостная хроматография | Распределительная жидкость - жидкостная хроматография |
По механизму взаимодействия сорбента и сорбата можно выделить несколько видов хроматографии:
-адсорбционная хроматография основана на различии в адсорбируемости веществ твердым сорбентом;
-распределительная хроматография - на различии в растворимости разделяемых веществ в неподвижной фазе (газовая хроматография) или на различии в растворимости веществ в подвижной и неподвижной жидких фазах;
-ионообменная хроматография - на разной способности веществ к ионному обмену;
-эксклюзионная хроматография - на различии в размерах и формах молекул разделяемых веществ;
-аффинная хроматография - на специфических взаимодействиях, характерныхдля некоторых биологических и биохимических процессов.
По технике выполнения различают
колоночную хроматографию (разделение проводится в
специальных колонках);
плоскостную хроматографию, когда разделение проводится на
специальной бумаге (бумажная хроматография) или в тонком слое сорбента (тонкослойная хроматография).
По цели хроматографирования выделяют
аналитическую хроматографию (качественный и количественный
анализ);
препаративную хроматографию (для получения веществ в чистом виде, для концентрирования и выделения микропримесей);
промышленную (производственную) хроматографию для
автоматического управления процессом.
По способу относительного перемещения фаз различают
фронтальную;
проявительную, или элюентную;
вытеснительную хроматографию.
В хроматографии подвижную фазу, вводимую в слой неподвижной фазы, называют элюентом, а подвижную фазу, вышедшую из колонки и содержащую разделенные компоненты, - элюатом.
Способы получения хроматограмм
Фронтальный метод
Это простейший по методике вариант хроматографии. Он состоит в том, что через колонку с адсорбентом непрерывно пропускают анализируемую смесь, например, компонентов А и В в растворителе. Вследствие сорбции веществ А и В сначала из колонки будет вытекать растворитель, затем растворитель и менее сорбирующийся компонент А, а затем и компонент В и, таким образом, через некоторое время состав раствора при прохождении через колонку меняться не будет. В растворе, вытекающем из колонки, определяют концентрацию каждого компонента и строят график в координатах концентрация вещества - объем раствора, прошедшего через колонку. Эту зависимость называют хроматограммой или выходной кривой (рис 1).
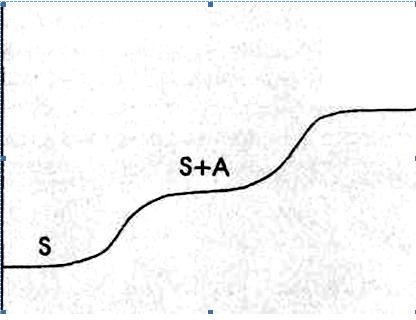
Рис. 1. Выходная кривая фронтального анализа.
При фронтальном способе получения хроматограммы в чистом виде можно выделить лишь одно вещество, поэтому фронтальный метод используется сравнительно редко. Данный метод применяется, например, для очистки раствора от примесей, если они сорбируются значительно лучше, чем основной компонент, или для выделения из смеси наиболее слабо сорбирующегося вещества.
Проявительный (элюентный) метод
При работе по этому методу в колонку вводят порцию анализируемой смеси, содержащей компоненты А и В в растворителе, и колонку непрерывно промывают газом-носителем или растворителем. При этом компоненты анализируемой смеси разделяются на зоны: хорошо сорбирующееся вещество В занимает верхнюю часть колонки, а менее сорбирующийся компонент А будет занимать нижнюю часть. Типичная выходная кривая изображена на
В газе или растворе, вытекающем из колонки, сначала появляется компонент В, далее — чистый растворитель, а затем компонент А. Чем больше концентрация компонента, тем выше пик и больше его площадь, что составляет основу количественного хроматографического анализа. Проявительный метод дает возможность разделять сложные смеси, он наиболее часто применяется на практике. Недостатком метода является уменьшение концентрации выходящих растворов за счет разбавления растворителем (газом — носителем).
Вытеснитепьный метод
В этом методе анализируемую смесь компонентов А и В в растворителе вводят в колонку и промывают раствором вещества С (вытеснитель), обладающим большей сорбируемостью, чем любое из разделяемых веществ. По мере продвижения по колонке элюент вытесняет вещество С, которое в свою очередь вытесняет вещество В, и т.д. Каждый из компонентов выделяется в чистом виде, но не количественно, так как зоны компонентов не разделены промежутками чистого сорбента (рис.3).
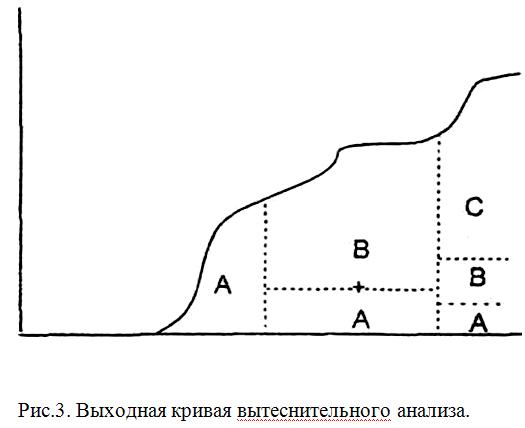
Концентрация раствора при хроматографировании не уменьшается в отличие от проявительного метода. Однако существенным недостатком вытеснительного метода является частое наложение зоны одного вещества на зону другого, поскольку зоны компонентов в этом методе не разделены зоной растворителя.
Выбор варианта хроматографического анализа
Для проведения хроматофафического анализа обычно используют следующую методику: выбирают нужные подвижные и неподвижные фазы в зависимости от свойств анализируемых веществ, т.е. выбирают определенный вариант хроматографии (рис.4), устанавливают необходимый режим хромотографии (рис.4), устанавливают необходимый режим хромотографии (температуру, скорость подачи подвижной фазы, детектор), затем проводят хроматографическое разделение и регистрируют сигнал. Используя полученный сигнал, определяют содержание каждого компонента в подвижной фазе после ее выхода из колонки
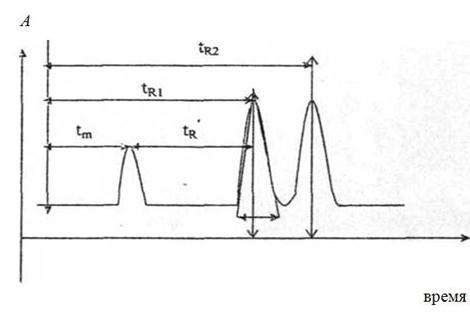
Рис.4 Схема выбора варианта хроматографического анализа
Хроматографические параметры
Наиболее важными хроматографическими параметрами, позволяющими оценить эффективность и селективность колонки и степень разделения различных веществ, являются: время удерживания, удерживаемый объем, коэффициент емкости, коэффициент удерживания, число теоретических тарелок, высота, эквивалентная теоретической тарелке, коэффициент селективности и коэффициент разделения.
На рис.5 представлена идеализированная хроматограмма смеси двух веществ. По оси абсцисс отложено время хроматографирования (можно отложить объем элюата), по оси ординат — аналитический сигнал, связанный с концентрацией веществ в элюате (отклик А). Рассмотрим основные хроматографические параметры, характеризующие поведение вещества в колонке. Время от момента ввода анализируемой пробы до момента регистрации максимума пика называют временем удерживания, или временем элюирования - (tR). Время удерживания складывается из двух составляющих — времени пребывания веществ в подвижной фазе (tm) и времени пребывания в неподвижной фазе (tS):
tR = tm+ tS
Значение tm фактически равно прохождения через колонку несорбируемого компонента. Значение tR не зависит от количества пробы, но зависит от природы вещества и сорбента и может меняться от колонки к колонке. Поэтому для характеристики истинной удерживающей способности следует ввести исправленное время удерживания (t'R): t'R = tR – tm
Часто для характеристики удерживания используют удерживаемый объем VR - объем подвижной фазы, который нужно пропустить через колонку с определенной скоростью, чтобы элюировать вещество:
VR = F tR
где F - объемная скорость потока, см З с-1.
Объем для вымывания несорбируемого компонента выражается через tm:
Vm = F xtm
Исправленный удерживаемый объем соответственно равен
V'R = VR – Vm
При постоянных условиях хроматографирования (скорость потока, давление, температура, состав фаз) значения tR и VR строго воспроизводимы и могут быть использованы для идентификации веществ.
Полезным в хроматографии является коэффициент удерживания (замедления) R — отношение скорости движения вещества к скорости движения растворителя:
Таким образом, R показывает, какую долю времени вещество находится в подвижной фазе.
Другой процесс распределения вещества между двумя фазами характеризуется коэффициентом распределения D. В данном случае
D=Cs/Cm
где Cm и Cs - равновесная концентрация вещества в подвижной и неподвижной фазах соответственно.
ОБРАБОТКА ХРОМАТОГРАФИЧЕСКИХ ДАННЫХ
Хроматография позволяет разделять не только компоненты смеси, но и определять ее качественный и количественный состав, поскольку положения хроматографического пика на хроматограмме (удерживаемый объем и время удерживания) для данной хромотографической системы характеризует природу данного вещества. Площадь, ограниченная этой кривой и нулевой линией детектора (хроматографический пик), пропорциональна количеству данного вещества, прошедшего через детектор.
Качественный анализ
Идентификация хроматографическими методами - это, прежде всего, идентификация по параметрам удерживания (tR, VR), поскольку они отличаются хорошей воспроизводимостью, относительные стандартные отклонения не превышают 2%. Совпадение величин удерживания неизвестного и стандартного соединения говорит о том, что эти соединения могут быть идентичными. Однако возможен случай, когда различные вещества имеют одинаковое время удерживания. Поэтому для большей достоверности идентификации сравнение хроматографических параметров известного и неизвестного вещества проводят в сильно различающихся условиях, например, получают данные об их хроматографическом поведении на колонках с различными неподвижными фазами. Если хроматографическое поведение стандартного и неизвестного вещества в таких случаях идентично, то достоверность идентификации возрастает до 99%.
При сравнении хроматограмм, полученных на разных приборах, чтобы избежать ошибок в идентификации, используют исправленное время удерживания и исправленный удерживаемый объем. Часто идентификацию проводят по относительному удерживанию (to™), т. е. по отношению удерживаемого объема определяемого компонента к удерживаемому объему вещества, принятого за стандарт:
tопт=t'R/tRcm=V'R/VRcm
Эта величина зависит только от состава подвижной и неподвижной фаз.
Существует еще один способ идентификации, основанный на одновременном использовании двух детекторов. Один детектор неспецифичен (катарометр, рефрактометр), а интенсивность сигнала другого детектора зависит от природы вещества, например, детектор -электронного захвата в газовой хроматографии (ГХ) или УФ детектор в жидкостной хроматографии. Сравнение хроматограмм, полученных с помощью двух детекторов, дает информацию, например, о составе и функциональных группах органических веществ.
Закономерность изменения удерживаемых объемов в гомологическом ряду органических соединений также создает основу для идентификации. Например, в ГХ используют зависимость логарифма исправленного объема от числа углеродных атомов в соединениях, принадлежащих к одному гомологическому ряду. Если установлено, что соединение относится к данному гомологическому ряду, его можно идентифицировать по графику зависимости логарифма исправленного объема от числа углеродных атомов. Для этого достаточно знать характеристики удерживания нескольких членов гомологического ряда.
Для качественной идентификации удобно пользоваться индексами удерживания Ковача (I), которые по существу являются относительными параметрами удерживания. В этом случае за стандарт берут два соседних алкана, один из которых элюируется до, а второй - после исследуемого соединения, т.е.
t'R(z) < t'R (x) < t'R(z + I)
где z - число атомов углерода в алкане.
Количественный анализ
Для проведения количественного анализа по хроматограмме сигнал детектора передается на электронное устройство, которое преобразует его в цифровую форму, либо на самописец с диаграммной лентой. В последнем случае количественный анализ проводят по высоте или площади пика, так как эти параметры пропорциональны концентрации или количеству вещества в хроматографической зоне
Измерение высот пиков проще, чем площадей, их можно измерить с большой точностью, особенно для веществ с малым временем удерживания. Чем меньше tR, тем уже, острее пик. Однако измерение площадей для количественного определения компонентов используется чаще. Для этого используют несколько способов (рис.8). Обычно проводят касательные к тылу и фронту пика и соединяют их линией параллельной нулевой линии. Площадь полученного треугольника составляет 96% от истинной и пропорциональна количеству вещества в пробе. Второй способ применяют при расчете площади только симметричных пиков. Для этого находят произведение высоты пика на его полуширину. Это произведение составляет 84% площади пика. Точность измерения площади пика зависит от отношения высоты пика к его ширине. Оптимальное значение лежит в пределах от 2 до 10.
Используя данные по высотам пиков или их площадям, можно рассчитать количественный состав пробы методами нормировки (с использованием или без использования поправочных коэффициентов), внешней (абсолютной калибровки) или внутренней стандартизации.
Метод нормировки чаще всего используют на практике (рис.9). Для его использования необходимо, чтобы на хроматограмме были зарегистрированы все компоненты, входящие в состав анализируемой смеси. Доля площади пика соответствует содержанию компонента в весовых процентах. При анализе смеси трех компонентов содержание компонента (%), например, соответствует пикуX на хроматограмме, можно рассчитать по формуле:
χ, % = (Sχ / (Sχ + Sy + Sz))·100,
где SX, Sy, Sz — площади пиков.
Эту формулу можно использовать только в том случае, если детектор одинаково чувствителен к каждому из разделяемых компонентов смеси, т.е. компоненты смеси, взятые в одинаковых количествах, дают одну и ту же площадь пика.
Если же чувствительность детектора различна по отношению к разным компонентам пробы, то используют поправочные коэффициенты fx, fy, fz учитывающие чувствительность детектора. Формула для расчета в этом случае записывается так:
χ, % = (Sχ fχ / ∑Sn fn) 100
Поправочные коэффициенты получают при анализе стандартных серий и рассчитывают по формуле:
fχ = (Scm Cχ / Sχ Ccm) fcm,
где Sx, Sст. — площади пиков анализируемого вещества и стандарта, Сх и С ст- — концентрации анализируемого веще-ства и стандарта, f ст. - поправочный коэффициент стандарта.
Метод внешнего стандарта используют при определении отдельных веществ или простых смесей, он удобен при определении микропримесей. Готовят два стандартных раствора определяемых компонентов, одинаковые их количества вводят в хроматограф и определяют площадь пика каждого компонента (S и S2). Результаты представляют либо графически (рис. 10), либо проводят расчет по формуле: X(%)=kSx
Градуировочный коэффициент К определяют при анализе проб стандартных серий смесей (рис. 9):
Рис. 9 Определение компонентов методом внешнего стандарта
Метод внутреннего стандарта применяют, если на хроматограмме отсутствуют пики некоторых компонентов анализи-руемой смеси. Метод основан на том, что в анализируемую смесь вводят некоторое количество стандартного вещества. Вещество, используемое в качестве внутреннего стандарта, должно удовлетворять ряду требований: должно полностью отделяться от других компонентов смеси; время удерживания его должно быть близким к tR определяемых компонентов; должно быть химически инертным и отсутствовать в определяемой пробе; его концентрация должна быть близкой к концентрации определяемых компонентов; пики симметричными. Для выполнения определения составляют смеси определенного точного состава внутреннего стандарта с каждым из компонентов, используют различные соотношения внутреннего стандарта и компонентов, получают хроматограммы таких смесей. Определяют площади пиков и для каждого компонента рассчитывают поправочный коэффициент по формуле:
k = (Sв.ст Сχ) /(Sχ Св.ст),
где Sв.ст, Sx — площади пиков внутреннего стандарта и определяемого компонента;
Св.ст, Сх- концентрации стандарта и исследуемого вещества в искусственных смесях.
Зная поправочные коэффициенты, расчет процентного содержания компонента проводят по формуле:
χ, % = kr (Sχ / Sв.ст)100,
где г =твст, /тпро6ы (m-масса, г).
внутреннего стандарта
Достоверность результатов и источники погрешностей. Систематические погрешности в хроматографический количествен-ный анализ вносят: подготовка и отбор представительной пробы, ее негомогенность, так как работают с малыми объемами проб; аппаратура — нелинейность детектора, различная его чувствительность к разным компонентам пробы; обработка хроматограмм. Чаще всего хроматографы, настроенные на работу в оптимальных условиях, не вносят значительного вклада в погрешность результата. Поэтому в общую дисперсию анализ включает дисперсию, связанную с измерением площади пика. Воспроизводимость измерения определения площадей пиков методом построения треугольника, выражаемая стандартным отклонением, составляет 4%, методом произведения высоты на ширину, измеренную на половине высоты - 2,5%, с помощью электронного интегратора - 0,4%.
|
|
|
|
Дата добавления: 2014-01-04; Просмотров: 6051; Нарушение авторских прав?; Мы поможем в написании вашей работы!