КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Методы предварительного восстановления
|
|
|
|
Методами окисления — восстановления можно определять любые окислители или восстановители. Описано применение для этой цели растворов перманганата, бихромата, бромата калия, иода. Используют и другие окислители, например Ce(SO4)2, NH4VO3. Известны и методы титрования окислителей восстановителями, например растворами FeSO-ь TiCl3 (титанометрия), SnCl2, CrCl2 (хромометрия). Эти методы
распространены мало, так как рабочие растворы сильных восстановителей необходимо защищать от окисления кислородом воздуха, сохраняя их и проводя титрование в атмосфере инертного газа, что требует применения специальных приспособлений. Кроме того, для фиксации конечной точки титрования нужны окислительно-восстановительные индикаторы, что не всегда удобно или доступно.
При титровании некоторыми окислителями нельзя обходиться без индикаторов, например при использовании рабочих растворов сульфата церия, ванадата аммония, бихрсмата калия. Поэтому наибольшее распространение в практике получили методы титрования окислителями, в частности растворами перманганата и иода; в первом случае индикатором служит сам рабочий раствор, во втором — специфический по отношению к иоду индикатор — крахмал. Высокая устойчивость растворов бихромата калия и возможность непосредственного использования его в качестве исходного вещества нередко делают применение К2С12О7 более удобным по сравнению с другими окислителями. Кроме того, потенциал бихромата калия ниже, чем у перманганата, вследствие чего титрование первым проходит без побочных реакций, мешающих определению. Бромат калия лучше всего применять в качестве титрднта для определения ряда органических веществ, с которыми он реагирует стехиометрически с образованием различных бромзамещенных соединений.
|
|
|
Несмотря на перечисленные достоинства, применение окислителей связано со следующими недостатками. Обычно предварительная подготовка пробы к анализу состоит в переведении анализируемого материала в раствор посредством обработки различными кислотами; чаще всего применяют азотную кислоту или ее смесь с хлороводородной или серной кислотой. Так, медные сплавы растворяют в азотной кислоте, причем содержащиеся в них элементы — железо, олово и другие — превращаются в соединения высших степеней окисления. При анализе различных чугунов и сталей необходимо определять ванадий, молибден, вольфрам, титан и некоторые другие легирующие элементы, которые вследствие обработки пробы окислительными агентами также содержатся в полученном растворе в высших степенях окисления. Железные руды содержат оксиды железа; растворяя их в хлороводородной кислоте с добавками различных окислителей, получают железо в степени окисления +3 и т. д.
Таким образом, титрование окислителями — КМ11О4, K2C12O7 и другими — возможно только после переведения определяемых элементов в низшие степени окисления. Восстановление необходимо провести количественно, для чего обычно вводят больший или меньший избыток восстановителя. Перед титрованием этот избыток необходимо каким-либо способом устранить, иначе рабочий раствор окислителя будет расходоваться не только на реакцию с определяемым элементом, но также на окисление восстановителя, и результат титрования окажется неправильным.
ТЕМА 11
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЙ
Лекция. Физико-химические и физические (инструментальные) методы анализа. Указанные методы анализа применяются в случае присутствия зависимости между измеряемыми физическими свойствами в-в и их качественным и количественным составом. Поскольку для измерения физических св-в в-в применяются различные приборы (инструменты), то эти методы наз-ся инструментальными. Классификация физических и физико-химических методов анализа. Основана на учете измеряемых физических и физико-химических св-в в-ва или изучаемой системы. Оптические методы основаны на измерении оптических св-в в-в. Хроматографические на использовании способности различных в-в к избирательной сорбции. Электрохимические методы основаны на измерении электрохимических св-в системы. Радиометрические основаны на измерении радиоактивных св-в в-в. Термические на измерении тепловых эффектов соответствующих процессов. Масс-спектрометрические на изучении ионизированных фрагментов («осколков») в-в. Ультразвуковые, магнитохимические, пикнометрические и т.д. Достоинства инструментальных методов анализа: низкий предел обнаружения 1 -10-9мкг; малая предельная концентрация, до 10-12г/мл определяемого в-ва; высокая чувствительность, формально определяемая величиной тангенса угла наклона соответствующей градуировочной кривой, отражающей графически зависимость измеряемого физического параметра, который откладывается обычно по оси ординат, от кол-ва или концентрации определяемого в-ва (ось абсцисс). Чем больше тангенс угла наклона кривой к оси абсцисс, тем чувствительнее метод, что означает следующее: для получения одинакового «отклика» - изменения физического свойства – требуется меньшее изменение концентрации или кол-ва измеряемого в-ва. К достоинствам относится высокая селективность (избирательность) методов, т. е. сотавные компоненты смесей можно определять без разделения и выделения этих компонентов; малая продолжительность времени проведения анализа, возможность их автоматизации и компьютеризации. Недостатки: сложность аппаратуры и высокая стоимость; большая погрешность (5 -20 %), чем в классическом химич-ом анализе (0,1 -0,5%); хуже воспроизводимость. Оптические методы анализа основаны на измерении оптических св-в в-ва (испускание, поглощение, рассеяние, отражение, преломление, поляризация света), проявляющихся при взаимодействии электромагнитного излучения с в-вом. Классификация по изучаемым объектам: атомный и молекулярный спектральный анализ. По характеру взаимодействия электромагнитного излучения с в-ом. При этом различают следующие методы. Атомно-абсорбционный анализ, в основе которого лежит измерениепоглощения монохроматического излучения атомами определяемого в-ва в газовой фазе после атомизации в-ва. Эмиссионный спектральный анализ – измерение интенсивности света, излучаемого в-ом (чаще всего атомами или ионами) при его энергетическом возбуждении, например, в плазме электрического разряда. Пламенная фотометрия – использование газового пламени в качестве источника энергетического возбуждения излучения. Нефелометрия – измерение рассеивания света частицами света дисперсной системы (среды). Турбидиметрический анализ – измерение ослабления интенсивности излучения при его прохождении через дисперсную среду. Рефрактометрический анализ измерение показателей светопреломления в-в. Поляриметрический анализ измерение величины оптического вращения – угла вращения плоскости поляризации света оптически активными в-ми. По области используемого электромагнитного спектра классифицируют следующие методы: спектроскопия (спектрофотометрия) в УВИ области спектра, т. е. в ближайшей ультрафиолетовой области спектра – в интервале длин волн 200 – 400 нм и в видимой области – в интервале длин волн 400 – 700 нм. Инфракрасная спектроскопия, изучающая участок электромагнитного спектра в интервале 0,76 – 1000 мкм (1 мкм=10-6 м), реже рентгеновская и микроволновая спектроскопия. По природе энергетических переходов в различных спектрах – электронных (изменение энергии электронных состояний атомов, ионов, радикалов, молекул, кристаллов в УВИ области); колебательных (при изменении энергии колебательных состояний 2-х и многоатомных ионов, радикалов, молекул, а также жидких и твердых фаз в ИК области); вращательных также в ИК и микроволновой области. Т.о. взаимодействие между молекулами и электромагнитным излучением заключается в том, что путем поглощения электромагнитного излучения молекулы переходят в возбужденное состояние. При этом важную роль играет энергия, т. е. длина волны поглощенного излучения. Так, в рентгеновских лучах, длина волны которых 0,05 – 5 нм, происходит процесс возбуждения внутренних электронов в атомах и молекулах; в ультрафиолетовых лучах (5 – 400 нм) происходит процесс возбуждения внешних электронов в атомах и молекулах; видимый свет (400 – 700 нм) происходит возбуждение внешних электронов в сопряженных π-электронных системах; инфракрасное излучение (700 нм – 500 мк) происходит процесс возбуждения колебаний молекул; микроволны (500 мк – 30 см) процесс возбуждения вращения молекул; радиоволны (более 30 см) процесс возбуждения спиновых переходов в атомных ядрах (ядерный магнитный резонанс). Поглощение излучений позволяет в спектрометрии их измерять и регистрировать. При этом падающее излучение делится на эталонное и измеряемое при одинаковой интенсивности. Измеряемое излучение проходит через пробу; при этом происходит поглощение, изменяется интенсивность. При поглощении энергии электромагнитного излучения частицы в-ва (атомы, молекулы, ионы) увеличивают свою энергию, т. е. переходят в более высоколежащее энергетическое состояние. Электронные, колебательные, вращательные энергетические состояния частиц в-ва могут изменяться лишь дискретно, на строго определенную величину. Для каждой частицы существует индивидуальный набор энергетических состояний – энергетических уровней (термов), например, электронных уровней энергии. Электронные энергетические уровни молекул и многоатомных ионов имеют тонкую структуру – колебательные подуровни; поэтому одновременно с чисто электронными переходами осуществляются и колебательные переходы. Каждому электронному (электронно-колебательному) переходу с нижнего энергетического уровня на более высоко лежащий электронный уровень отвечает полоса в электронном спектре поглощения. Так как разность между электронными уровнями для каждой частицы (атома, иона, молекулы) строго определенна, то строго определенным является и положение полосы в электронном спектре поглощения, соответствующей тому или иному электронному переходу, т. е. длина волны (частота, волновое число) максимума полосы поглощения. Различия в интенсивности измеряются детектором и записываются на самописце в виде сигнала (пика), стр 318, химия, справочник школьника и студента, схема спектрометра. Ультрафиолетовая спектроскопия и абсорбционная спектроскопия в видимой области. Поглощение электромагнитного излучения из ультрафиолетовой и видимой части спектра; возбуждает переходы электронов в молекулах с занятых на незанятые энергетические уровни. Чем больше разность в энергии между энергетическими уровнями, тем большую энергию, т.е. более короткую длину волны, должно иметь излучение. Часть молекулы, которая в значительной части определяет поглощение света, называется хромофором (буквально, несущие цвет)– это атомные группы, влияющие на поглощение света молекулой, в особенности сопряженные и ароматические системы π-электронов. Структурные элементы хромофоров в основном и участвуют в поглощении кванта световой энергии, что приводит к появлению полос в сравнительно узком участке спектра поглощения соединений. Практическое значение для определения строения органических молекул имеет область от 200 до 700 нм. Количественное измерение: наряду с положением максимума поглощения для анализа важно значение экстинкции (ослабления) излучения, т. е. интенсивности его поглощения. В соответствии с законом Ламберта – Бера Е=lgI0 /I=εcd, Е – экстинкция, I0 – интенсивность падающего света, I – интенсивность проходящего света, ε – молярный коэффициент экстинкции, см2/моль, c – концентрация, моль/л, d – толщина слоя пробы, см. Экстинкция зависит от концентрации поглощающего в-ва. Методы абсорбционного анализа колориметрия, фотоэлектроколориметрия, спектрометрия. Колориметрия самый простой и старый метод анализа, основан на визуальном сравнении окраски жидкостей (определение рН почвы на приборе Алямовского) – самый простой метод сравнения с серией эталонных р-ов. Широко распространены 3-и метода колориметрии: метод стандартных серий (метод шкалы), метод уравнивания окрасок и метод разбавления. Используются стеклянные колориметрические пробирки, стеклянные бюретки, колориметры, фотометры. Метод шкалы – это определение рН на приборе Алямовского, т. е. серия пробирок с различной конц-ей в-ва и разная по изменению интенсивности цвета р-ра или эталонных р-ов. Фотоколориметрия – метод основан на измерении интенсивности немонохроматического светового потока, прошедшего через анализируемый р-р с помощью фотоэлементов. Световой поток от источника излучения (лампы накаливания) проходит через светофильтр, пропускающий излучение лишь в определенном интервале длин волн, через кювету с анализируемым р-ом и попадает на фотоэлемент, преобразующий световую энергию в фототок, регистрируемый соответствующим прибором. Чем больше светопоглощение анализируемого р-ра (т. е. чем выше его оптическая плотность), тем меньше энергия светового потока, попадающего на фотоэлемент. ФЭКи снабжаются н-ми светофильтрами, имеющими максимум светопропускания при различных длинах волн. При наличии 2-х фотоэлементов происходит измерение 2-х световых потоков, одного через анализируемый р-р, другого через р-р сравнения. Концентрацию исследуемого в-ва находят по градуировочному графику.
|
|
|
|
|
|
|
|
|
Электрохимические методы анализа основаны на электродных реакциях и на переносе электричества через р-ры. В количественном анализе используется зависимость величин измеряемых параметров электрохимических процессов (разность электрических потенциалов, ток, кол-во электричества) от сод-ия определяемого в-ва в р-ре, участвующего в данном электрохимическом процессе. Электрохимические процессы – такие процессы, которые сопровождаются одновременным протеканием химических реакций и изменением электрических св-в системы, которую в подобных случаях можно наз-ать электрохимической системой. Основные принципы потенциометрии
Как следует из названия метода – в нем измеряется потенциал. Для пояснения, что за потенциал и почему он возникает, рассмотрим систему состоящую из металлической пластины и находящегося с ней в контакте раствора, содержащего ионы того же металла (электролит) (рис. 1). Такая система называется электродом. Любая система стремится к такому состоянию, которое отвечает минимуму ее внутренней энергии. Поэтому в первый момент после погружения металла в раствор на границе раздела фаз начинают протекать процессы, ведущие к снижению внутренней энергии системы. Предположим, что ионизированное состояние атома металла энергетически более «выгодно», чем нейтральное (возможен и обратный вариант). Тогда в первый момент времени атомы металла будут переходить из поверхностного слоя пластины в раствор, оставляя в ней свои валентные электроны. При этом поверхность пластины приобретает отрицательный заряд, причем этот заряд растет по мере увеличения количества атомов металла, перешедших в виде ионов в раствор. Электростатические силы притяжения разноименных зарядов (отрицательнозаряженные электроны в пластине и положительные ионы металла в растворе) не позволяют удалиться этим зарядам от границы раздела фаз, а также вызывают обратный процесс перехода ионов металла из раствора в металлическую фазу и восстановления их там. Когда скорости прямого и обратного процессов становятся одинаковыми, наступает равновесие. Состояние равновесия системы характеризуется разделением зарядов на границе раздела фаз, т. е. появляется «скачок» потенциала. Следует отметить, что описанный механизм возникновения электродного потенциала является не единственным, в реальных системах протекает также множество других процессов, приводящих к образованию «скачка» потенциалов на межфазовой границе. Кроме того, «скачок» потенциала может возникать на границе раздела фаз не только при контакте электролита с металлом, но и при контакте электролита с другими материалами, например, полупроводниками, ионообменными смолами, стеклами и т. д. При этом ионы, концентрация которых влияет на потенциал электрода называются потенциалопределяющими. Потенциал электрода зависит от природы материала, контактирующего с электролитом, концентрации потенциалопределяющих ионов в растворе и температуры. Этот потенциал измеряется относительно другого электрода, потенциал которого постоянен. Т. о., установив эту связь, возможно использовать ее в аналитической практике для определения концентрации ионов в растворе. При этом электрод, потенциал которого измеряется, носит название измерительный, а электрод, относительно которого производятся измерения – вспомогательный или электрод сравнения. Постоянство потенциала электродов сравнения достигается постоянством концентрации потенциалопределяющих ионов в его электролите (электролит №1). Состав электролита №2 может меняться. Для предотвращения смешивания двух разных электролитов они разделяются мембраной, проницаемой для ионов. Потенциал измерительного электрода принимается равным измеренной э.д.с., приведенной электрохимической системы. Применяя в качестве электролита №2, растворы известного состава можно установить зависимость потенциала измерительного электрода от концентрации потенциалоопределяющих ионов. Эта зависимость в дальнейшем может быть использована при анализе раствора неизвестной концентрации.
Для стандартизации шкалы потенциалов в качестве электрода сравнения принят стандартный водородный электрод, потенциал которого принят равным нулю при любой температуре. Однако при обычных измерениях водородный электрод применяется редко из-за своей громоздскости. В повседневной практике применяют другие более простые электроды сравнения, потенциал которых относительно водородного электрода определен. Поэтому, при необходимости, результат измерения потенциала, проведенного относительно таких электродов, может быть пересчитан относительно водородного электрода. Наиболее широко распространенными являются хлорсеребряный и каломельный электроды сравнения. Разность потенциалов измерительного электрода и электрода сравнения является мерой концентрации определяемых ионов. Электродную функцию можно описать с помощью линейного уравнения Нернста: Е = Е0 + 2,3 RT/nF *lg а, где Е – разность потенциалов между измерительным электродом и электродом сравнения, мВ; Е0 – константа, зависящая в основном от свойств электрода сравнения (стандартный потенциал электрода), мВ; R – газовая постоянная, Дж*моль-1* К-1.; n – заряд иона с учетом его знака; F – число Фарадея, Кл/моль; Т – абсолютная температура, 0 К; член 2,3 RT/nF, входящий в уравнение Нернста при 250 С равен 59,16 мВ для однозарядных ионов. Метод без наложения внешнего (постороннего) потенциала классифицируется как метод, основанный на учете природы источника электрической энергии в системе. В этом методе источником эл.эн. служит сама элек-хим-ая система, представляющая собой гальванический элемент (гальваническую цепь) – потенциометрические методы. ЭДС и электродные потенциалы в такой системе зависят от сод-ия определяемого в-ва в р-ре. Электрохимическая ячейка включает 2-ва электрода – индикаторный и электрод сравнения. Величина ЭДС, генерируемой в ячейке, равна разности потенциалов этих 2-х электродов. Потенциал электрода сравнения в условиях проведения потенциометрического определения остается постоянным, то ЭДС зависит только от потенциала индикаторного электрода, т. е. от активностей (концентраций) тех или иных ионов в р-ре. На этом и основано потенциометрическое определение концентрации данного в-ва в анал-ом р-ре. Применяют как прямую потенциометрию, так и метод потенциометрического титрования. При определении рН р-ов в кач-ве индикаторных используются электроды потенциал которых зависит от конц-ии ионов водорода: стеклянный, водородный, хингидронный (окислительно-восстановительный электрод в виде платиновой проволоки, погруженной в р-р НС1, насыщенной хингидроном – эквимолекулярным соединением хинона с гидрохиноном) и нек-ые др. Мембранные или ион-селективные электроды имеют реальный потенциал, зависящий от активности тех ионов в р-ре, кот-ые сорбируются мембраной электрода (твердой или жидкой) метод наз-ся ионометрией. Спектрофотометрами наз-ют приборы, позволяющие производить измерения светопоглощения образцов в узких по спектральному составу пучках света (монохроматический свет). Спектрофотметры позволяют разлагать белый свет в непрерывный спектр, выделять из этого спектра узкий интервал длин волн (1 – 20 нм ширина выделяемой полосы спектра), пропускать изолированный пучок света через анализируемый р-р и измерять с высокой точностью интенсивность этого пучка. Поглощение света окрашенным в-ом в р-ре измеряют, сравнивая его с поглощением нулевого р-ра. В спектрофотометре сочетаются два прибора: монохроматор для получения монохроматического светового потока и фотоэлектрический фотометр, предназначенный для измерения интенсивности света. Монохроматор состоит из источника света, диспергирующего устройства (разлагающего белый свет в спектр) и устройства регулирующего величину интервала длин волн светового пучка, падающего на р-р.
Из разнообразных физико-химических и физических методов анализа наибольшее значение имеют 2-ве группы методов: 1 – методы, основанные на изучении спектральных характеристик в-ва; 2 – методы, основанные на изучении физико-химических параметров. Спектральные методы основаны на явлениях, происходящих при взаимодействии вещества с различными видами энергии (электромагнитным излучением, термической энергией, электрической и пр.). К основным видам взаимодействия в-ва с лучистой энергией относится поглощение и испускание (эмиссия) излучения. Характер явлений, обусловленных поглощением или испусканием, в принципе одинаков. При взаимодействии излучения с в-вом частицы его (атомы молекулы) переходят в возбужденное состояние. Через некоторое время (10-8 с) частицы возвращаются в основное состояние, испуская избыточную энергию в виде электромагнитного излучения. Эти процессы связаны с электронными переходами в атоме или молекуле.
Электромагнитное излучение можно охарактеризовать длиной волныλ или частотой ν, которые связаны между собой соотношением ν=с/λ, где с – скорость света в вакууме (2,298108 м/с). Совокупность всех длин волн (частот) электромагнитного излучения составляет электромагнитный спектр от γ-лучей (коротковолновая область, фотоны обладают высокой энергией) до видимой области спектра (400 – 700 нм) и радиоволн (длинноволновая область, фотоны с низкой энергией).
На практике имеют дело с излучением, характеризующимся определенным интервалом длин волн (частот), т. е. с определенным участком спектра (или, как говорят, с полосой излучения). Часто для аналитических целей используется и монохроматический свет (световой поток, в котором электромагнитные волны имеют одну длину волны). Избирательное поглощение атомами и молекулами излучения с определенными длинами волн приводит к тому, что каждое в-во характеризуется индивидуальными спектральными характеристиками.
Для аналитических целей используют как поглощение излучения атомами и молекулами (соответственно атомно- абсорбционная спектроскопия), так и испускание излучения атомами и молекулами (эмиссионная спектроскопия и люминесценция).
Спектрофотометрия основана на избирательном поглощении электромагнитного излучения в-вом. Измеряя поглощение в-вом излучения различных длин волн, можно получить спектр поглощения, т. е. зависимость поглощения от длины волны падающего света. Спектр поглощения – это качественная характеристика в-ва. Количественной характеристикой является количество поглощенной энергии или оптическая плотность раствора, которая зависит от концентрации поглощающего в-ва по закону Бугера-Ламберта-Бера: D=εІс, где D – оптическая плотность, i – толщина слоя; с – концентрация, моль/л; ε – молярный коэффициент поглощения (ε = D при І=1 см и с=1 моль/л). Величина ε служит характеристикой чувствительности: чем больше значение ε, тем меньшие количества в-ва можно определить. Многие в-ва (особенно органические) интенсивно поглощают излучение в УФ- и видимой областях, что делает возможным их непосредственное определение. Большинство ионов, наоборот, слабо поглощают излучение видимой области спектра (ε ≈ 10…1000), поэтому их обычно переводят в другие, более интенсивно поглощающие соединения, а затем проводят измерения. Для измерения поглощения (оптической плотности) используют спектральные приборы 2-х видов: фотоэлектроколориметры (с грубой монохроматизацией) и спектрофотометры (с более тонкой монохроматизацией). Наиболее распространенным является фотометрический метод анализа, количественные определения в котором основаны на законе Бугера-Ламберта-Бера. Основными приемами фотометрических измерений являются: метод молярного коэффициента светопоглощения, метод градуировочного графика, метод стандартов (метод сравнения), метод добавок. В методе молярного коэффициента светопоглощения измеряют оптическую плотность D исследуемого р-ра и по известному значению молярного коэффициента светопоглощения ε рассчитывают концентрацию с поглощающего в-ва в растворе: с = D/(ε І). В методе градуировочного графика готовят ряд стандартных растворов с известным значением концентрации с определяемого компонента и определяют их значение оптической плотности D. По полученным данным строят градуировочный график – зависимость оптической плотности раствора от концентрации в-ва: D = f(с). В соответствии с законом Бухера-Ламберта-Бера график представляет собой прямую линию. Затем измеряют оптическую плотность D исследуемого раствора и по градуировочному графику определяют концентрацию определяемого соединения. Метод сравнения (стандартов) основан на сравнении оптической плотности стандартного и исследуемого растворов: Dст=ε*І*сст и Dх= ε*І*сх, откуда Dх/ Dст=ε*І*сх/ε*І*сст и сх=сст*Dх/Dст. В методе добавок сравниваются значения оптической плотности исследуемого раствора и того же раствора с добавлением (са) известного количества определяемого компонента. По результатам определений рассчитывают концентрацию в-ва в исследуемом растворе: Dх= ε*І*сх и Dх+а= ε*І*(сх+са), откуда Dх/Dх+а= ε*І*сх/ε*І*(сх +са) и сх=са* Dх/Dх+а – Dх..
Атомно-абсорбционная спектроскопия основана на избирательном поглощении излучения атомами. Для переведения вещества в атомарное состояние раствор образца впрыскивают в пламя или подогревают в специальной кювете. В результате растворитель улетучивается или сгорает, а твердое в-во атомизируется. Большая часть атомов остается в невозбужденном состоянии, и лишь небольшая часть возбуждается с последующим испусканием излучения. Набор линий, соответствующий длинам волн поглощаемого излучения, т. е. спектр, является качественной характеристикой, а интенсивность этих линий – соответственно количественной характеристикой в-ва.
Атомно-эмиссионная спектроскопия основана на измерении интенсивности света, излучаемого возбужденными атомами. Источниками возбуждения могут быть пламя, искровый разряд, электрическая дуга и др. Для получения спектров испускания пробу в виде порошка или раствора вводят в источник возбуждения, где происходит переход в-ва в газообразное состояние или частичный распад его на атомы и простые (по составу) молекулы. Качественной характеристикой в-ва является его спектр (т. е. набор линий в спектре испускания), а количественной – интенсивность этих линий.
Люминесценция основана на испускании излучения возбужденными молекулами (атомами, ионами) при переходе их в основное состояние. Источниками возбуждения при этом могут быть ультрафиолетовое и видимое излучение, катодные лучи, энергия химической реакции и пр. Энергия излучения (люминесценции) всегда меньше поглощенной энергии, т. к. часть поглощенной энергии еще до начала испускания преобразуется в тепловую. Следовательно, люминесцентное испускание всегда имеет меньшую длину волны, чем длина волны поглощенного при возбуждении света. Люминесценция может использоваться как для обнаружения в-в (по длине волны), так и для их количественного определения (по интенсивности излучения). Электрохимические методы анализа основаны на взаимодействии в-ва с электрическим током. Протекающие при этом процессы локализованы либо на электродах, либо в приэлектродном пространстве. Большинство методов относятся к первому из этих типов. Потенциометрия. Электродным процессом называется гетерогенная реакция, при которой заряженная частица (ион, электрон) переносится через границу раздела фаз. В рез-те такого переноса на пов-ти электрода возникает разность потенциалов, обусловленная образованием двойного электрического слоя. Как всякий процесс, электродная реакция с течением времени приходит к равновесию, и на электроде устанавливается равновесный потенциал. Измерение величин равновесных электродных потенциалов является задачей потенциометрического метода анализа. Измерения при этом проводят в электрохимической ячейке состоящей из 2-х полуэлементов. Одиг из них содержит индикаторный электрод (потенциал которого зависит от концентрации определяемых ионов в растворе в соответствии с уравнением Нернста), а другой – электрод сравнения (потенциал которого постоянен и не зависит от состава раствора). Метод может быть реализован в варианте прямой потенциометрии или в варианте потенциометрического титрования. В первом случае измеряют потенциал индикаторного электрода в анализируемом растворе относительно электрода сравнения и по уравнению Нернста рассчитывают концентрацию определяемого иона. В варианте потенциометрического титрования определяемый ион титруют подходящим реагентом, следя одновременно за изменением потенциала индикаторного электрода. По полученным данным строят кривую титрования (зависимость потенциала индикаторного электрода от объема прибавленного титранта). На кривой вблизи точки эквивалентности наблюдается резкое изменение значения потенциала (скачок потенциала) индикаторного электрода, что позволяет рассчитать содержание определяемого иона в растворе. Электродные процессы очень многообразны. В целом их можно классифицировать на 2-ве большие группы: процессы, происходящие с переносом электронов (т. е. собственно электрохимические процессы), и процессы, связанные с переносом ионов(при этом электроду присуща ионная проводимость). В последнем случае речь идет о так называемых ионселективных мембранных электродах, широко применяемых в настоящее время. Потенциал такого электрода в растворе, содержащем определяемые ионы, зависит от их концентрации по уравнению Нернста. К этому же типу электродов относится и стеклянный электрод, применяемый в рН-метрии. Возможность создания большого числа мембранных электродов с высокой селективностью к тем или иным ионам выделила эту область потенциометрического анализа в самостоятельную отрасль – ионометрию.
Полярография. При прохождении тока в электрохимической ячейке наблюдается отклонение величин электродных потенциалов от их равновесных значений. В силу ряда причин возникает так называемая электродная поляризация. Явление поляризации, возникающей в процессе электролиза на электроде с малой поверхностью, лежит в основе данного метода анализа. В этом методе к электродам, опущенным в исследуемый раствор, прикладывают возрастающую разность потенциалов. При малой величине разности потенциалов ток через раствор практически не идет (т. н. остаточный ток). При увеличении разности потенциалов до величины, достаточной для разложения электролита, сила тока резко возрастает. Эту величину разности потенциалов называют потенциалом разложения. Измеряя зависимость силы тока, проходящего через раствор, от величины приложенного напряжения, можно построить т. н. вольтамперную кривую, которая позволяет с достаточной точностью определить качественный и количественный состав раствора. При этом качественной характеристикой в-ва является величина разности потенциалов, достаточная для его электрохимического разложения (потенциал полуволны ЕS), а количественной – величина прироста силы тока, обусловленная его электрохимическим разложением в растворе (высота длины волны Н, или различие в величинах предельного диффузионного тока и остаточного тока). Для количественного определения концентрации в-ва в растворе используют следующие приемы: метод градуировочного графика, метод стандартов, метод добавок. Кондуктометрический метод анализа основан на зависимости электропроводности раствора от концентрации электролита. Применяется, как правило, в варианте кондуктометрического титрования, точку эквивалентности в котором определяют по перегибу кривой титрования (зависимости электропроводности от количества прибавленного титранта). Амперометрическое титрование является разновидностью потенциометрического титрования, только индикаторным электродом является полярографическое устройство, т.е. применяется микроэлектрод с наложенным напряжением.
СОДЕРЖАНИЕ ДИСЦИПЛИНЫ
ЛЕКЦИОННЫЙ КУРС
Тема 1. Введение. Предмет и задачи аналитической химии. Качественный анализ.
Краткий очерк развития аналитической химии. Понятие о чистых веществах и смесях. Химические свойства веществ. Понятие анализа и синтеза. Теоретические основы химического анализа. Методы аналитической химии: сухой, мокрый, физический, физико-химический, химические. Характеристика методов аналитической химии в зависимости от количества исследуемого вещества, объема и техники выполняемых операций. Аналитические реакции и требования, предъявляемые к ним. Методы обнаружения и разделения элементов – качественный анализ, качественные реакции. Дробный и систематический анализ. Аналитические классификации катионов, анионов. Примеры решения аналитических задач.
Тема 2. Закон действующих масс. Химическое равновесие.
Кинетика химических реакций. Понятие химической системы, фазы, процесса. Идеальные и реальные системы. Теоретические положения и математическое выражение закона действующих масс. Химическое равновесие, константа химического равновесия. Принцип Ле-Шателье. Равновесие в гетерогенных системах. Растворы. Понятие растворимости. Насыщенные, пресыщенные растворы. Произведение растворимости. Примеры решения аналитических задач.
Тема 3. Теория электролитической диссоциации.
Электролиты. Теория электролитической диссоциации С. Аррениуса. Количественные характеристики процесса электролитической диссоциации: константа диссоциации, степень диссоциации, ионная сила раствора, закон разбавления Оствальда. Понятие активности, коэффициента активности. Сильные и слабые электролиты. Диссоциация кислот, оснований, солей. Амфотерные соединения. Протолитическая теория Й. Н. Бренстеда и Т. М. Лоури. Факторы, влияющие на степень электролитической диссоциации. Солевой эффект и его значение в аналитической химии. Примеры решения аналитических задач.
Тема 4. Водородный и гидроксильный показатели. Буферные системы.
Автопротолиз. Ионное произведение воды. Водородный показатель – рН раствора. Шкала рН. Гидроксильный показатель, примеры вычислений. Методы определения рН среды: потенциометрический, колориметрический. Индикаторы. Действие одноименных ионов на степень диссоциации слабого электролита. Буферное действие, буферные растворы, буферная емкость. Амфотерность в качественном химическом анализе.
Тема 5. Гидролиз солей.
УЧЕБНО-МЕТОДИЧЕСКАЯ КАРТА
| № | Название темы | Наименование вопросов, которые изучаются | Ле кц ии | Ла бо рат орн ые | Нагл ядн ые посо бия | Самос тояте льная работа студен тов | Фор ма конт роля |
| Предмет, задачи и методы качественного анализа | Методологические основы аналитической химии. Виды качественных анализов. Дробный и систематический анализ. Системы качественного анализа катионов. Первая аналитическая группа катионов в соответствии с кислотно-основной классификацией катионов. Частные реакции катионов первой и второй аналитических групп катионов. Ход анализа смеси катионов первой и второй аналитических групп. | Табли цы, реак тивы, мето дичес кие инст рукц ии, ла бора торная посуда обору дован. | Прове дение качест венных хими ческих реакций по опре делению NН4+, К+, Nа+, Мg2+ Ва2+, Са2+, Sr2. | Прове рка оформ ления запи сей прак тичес кой рабо ты в рабоч их журна лах | |||
| Закон действующих масс | Закон действующих масс как теоретическая основа качественного анализа при изучении гетерогенных процессов. Третья и четвертая аналитические группы катионов: Аg+, Нg2+2, Рb2+, А13+, Zn2+, Сr3+, Sn2+, Sn4+. Анализ смеси катионов четырех аналитических групп. Гидролиз солей | Лабора торная посуда и обо рудова ние, реакти вы, ме тодиче ские инстру кции | Прове дение группо вых и ча стных реакций 3 и 4-ой аналити ческих групп катио нов | Инди виду альн ый опрос, реше ние задач, уравне ния гидро лиза солей | |||
| Окислительно-восстановительные процессы в качественном анализе | Правила определения степени окисления. Химические уравнения окисления-восстановления. Уравнение Нернста. Пятая и шестая аналитические группы катионов: Fе2+, Fе3+, Мn2+, Вi3+ , Сu2+, Нg2+, Сd2+, Ni2+, Со2+. | Лабора торная посуда и обо рудова ние, реакти вы, ме тодиче ские инстру кции | Проведе ние группо вых и частных реакций 5 и 6-ой аналити ческих групп и смеси катио нов | Конт роль ная рабо та, реше ние задач | |||
| Комплексообразование в качественном анализе | Образование комплексных соединений при проведении систематического и дробного анализа. Три аналитические группы анионов. Анализ раствора смеси неизвестных катионов и анионов | Лабора торная посуда и обо рудова ние, реакти вы, ме тодиче ские инстру кции | Проведе ние реакций трех аналити ческих групп анионов и раство ра неизвес тных ионов | Прове рка оформ ления запи сей прак тичес ких работ в рабо чих журна лах | |||
| Теория электролитической диссоциации. Буферные растворы | Коллоквиум по качественному анализу | Лабора торная посуда и обо рудова ние, реакти вы, ме тодиче ские инстру кции | Ответы на вопросы | Оценка колок виума | |||
| Количественный анализ, задачи и методы | Теоретические основы количественного анализа. Концентрация растворов и способы выражения концентрации растворов. Решение задач. | ||||||
| Гравиметрический (весовой) метод анализа | Сущность гравиметрического метода анализа. Осаждаемая и гравиметрическая формы осадка. Определение влажности пищевой продукции. | Анали тическ ие весы, сушиль ный шкаф, хими ческая посуда | Освое ние правил взвеши вания на ана литиче ских весах | Прове рка расче тов и запи сей в рабо чих журна лах | |||
| Титриметрический (объемный) анализ | Стандартные растворы, мерная посуда. Правила титрования. Принцип эквивалентности. Построение кривых титрования. Правила выбора индикатора | Реакти вы, ин дикато ры, ла бора торная посуда | Освоен ие прав ил титр ования, построе ние кри вых | Опрос, провер ка реше ния задач | |||
| Методы кислотно-основного титрования | Ацидиметрия, алкалиметрия. Индикаторы в кислотно-основном титровании. Определение временной карбонатной жесткости воды | Реакти вы, ин дикато ры, ла бора торная посуда | Опреде ление времен ной жестко сти воды | Провер ка расч етов, задач, фрон таль ный опрос | |||
| Методы осаждения и комплексонометрии | Классификация, характеристика методов осадительного титрования. Теоретические основы комплексонометрии. Определение постоянной жесткости воды комплексонометрическим методом | Реакти вы, ин дикато ры, ла бора торная посуда | Опреде ление постоян ной жестко сти воды | Инди видуа льный опрос, контр. работа | |||
| Физико-химические методы анализа | Колориметрические и спектрометрические методы анализа, потенциометрия. Калибровочные графики. Определение концентрации железа фотоколориметрическим методом. Определение рН раствора потенциометрическим методом | Фотоэл ектро колори метр, потен цио метр, реакти вы, ла бора торная посуда | Постро ение калибро вочного графика, работа на ФЭК, потен циомет ре | Конт роль ная работа |
ЛИТЕРАТУРА
Основная
1. Артеменко А. И. Справочное руководство по химии: Справ. пособие/ А. И. Артеменко, И. В. Тикунова, В. А. Малеванный – 2-е изд., перераб. и доп – М.: Высш. шк., 2002. – 367 с.
2. Васильев В. П. Аналитическая химия. В 2-х частях. Учеб. для химико-технол. спец. вузов. – М.: Высшая школа, 1989. – Ч. 1. – 320 с., Ч. 2. – 260 с.
3. Гельфман М. И., Юстратов В. П. Химия. 3-е изд., стер. Учеб. для вузов. – СПб.: Изд. «Лань», 2003. – 480 с.
4. Коровин Н. В., Мингулина Э. И., Рыжова Н. Г. Лабораторные работы по химии: Учеб. пособие для техн. направ. и спец. вузов / Под ред. Н. В. Коровина – 3-е изд., испр. – М.: Высш. шк., 2001. – 256 с.
5. Харитонов Ю. Я. Аналитическая химия (аналитика). В 2-х кн. Кн.1. Общие теоретические основы. Качественный анализ. Учеб. для вузов – М.: Высш. шк., 2001. – 615 с
6. Харитонов Ю. Я. Аналитическая химия (аналитика). В 2-х кн. Кн. 2. Количественный анализ. Физико-химические (инструментальные) методы анализа.
Учеб. для вузов – М.: Высш. шк., 2001. – 559 с.
7. Цитович И. К. Курс аналитической химии. Учебник. 7-ое изд., стер. – СПб.: Издательство «Лань», 2004. – 496 с.
Дополнительная
1. Барковский В. Ф., Горелик С. М., Городенцева Т. Б. Физико-химические методы анализа. Учебник для техникумов. – М.: Высш. шк., 1972. – 344 с.
2. Волков В. А. Выдающиеся химики мира: Биографический справочник. / Волков В. А., Вонский Е. В., Кузнецова Г. И.; Под ред. В. И. Кузнецова. – М.: Высш шк., 1991 – 656 с.
3. Гузей Л. С. Кузнецов В. Н. Новый справочник по химии: Справочник для школьников и абитуриентов/ Л. С. Гузей, В. Н. Кузнецов. Под ред. проф. С. Ф. Дунаева. – 1-е изд. – М.: Большая Медведица, 1998. – 354 с.
4. Зоммер К. и др. Химия: Справочник школьника и студента / К. Зоммер, К. Х. Вюнш, М. Цеттлер; Пер. с нем. под ред. проф. Р. А. Лидина. – 2-ое изд.,стереотип. – М.: Дрофа, 2000. – 384 с.
5. Соколова И. Ф. Химия. Пособие для старшеклассников и абитуриентов химических и медицинских вузов. – М.: Изд-во «Московский лицей», 2001. – 272 с.
6. Фигуровский Н. А. История химии: Учеб. пособие для студентов пед. ин-тов по хим. и биол. спец. – М.: Просвещение, 1979. – 311 с
Условия образования осадков.
1. Осаждение и концентрация ионов. Осадок малорастворимого электролита образуется только тогда, когда произведение концентраций ионов превысит величину его произведения растворимости (при определенной температуре). Известно, что не существует веществ, абсолютно не растворимых в воде. Поэтому при осаждении в растворе всегда остается какое-то количество ионов, отвечающее величине произведения растворимости электролита, если оно не очень велико (менее 10-5 моль/л) и выполнению дальнейших аналитических операций не мешает, то говорят, что достигнуто практически полное осаждение. Но часто для полного осаждения требуется соблюдение ряда условий, так как полнота осаждения зависит от растворимости осаждаемого соединения, количества реактива-осадителя, его степени диссоциации, гидролизуемости, рН раствора и других факторов (стр. 3, растворимости и стр. 20 печатн.) Осаждение и растворимость. При прочих равных условиях осаждение будет тем полнее, чем менее растворимо соединение, в виде которого мы осаждаем данный ион. По произведениям растворимости видно, что из солей бария наименее растворим сульфат бария ПР его равно 1,110-10, а из солей кальция оксалат кальция – 2,610-9. Для осаждения катионов бария следует брать сульфат ионы, а кальция оксалат ионы. Осаждение и количество осадителя. Для практически полного осаждения иона в виде его соединения, заметно растворимого в воде, нужно добавлять избыток осаждающего реактива (полуторный избыток осадителя или несколько меньший).
Цель работы — ознакомление с электрохимическим методом определения степени диссоциации электролитов, основанном на свойстве их водных растворов проводить электрический ток. Изучение зависимости степени диссоциации слабого электролита от концентрации раствора.
ТЕМА: ТИТРИМЕТРИЧЕСКИЙ (ОБЪЕМНЫЙ) АНАЛИЗ. ТИТРОВАНИЕ РАСТВОРА ЩЕЛОЧИ РАСТВОРОМ КИСЛОТЫ.
Получите у лаборанта в пронумерованной мерной колбе щелочь и приготовьте ее водный раствор в объеме данной колбы. Отберите пипеткой приготовленный раствор в коническую колбу и добавьте 1-2 капли фенолфталеина.
В бюретку налейте раствор соляной кислоты известной концентрации и приведите бюретку в рабочее положение.
Оттитруйте отобранный раствор щелочи раствором НС1. Конец титрования определяют по исчезновению окраски фенолфталеина. Титрование проведите не менее двух раз. Если результаты титрования одинаковы или отличаются друг от друга не более чем на 0,1 мл, закончите титрование. В противном случае титрование повторите. Полученные данные сведите в таблицу:
| Номер колбы | Объем пипетки, мл | с (НС1), моль/л | V НС1, мл | ||
| среднее | |||||
Контрольные вопросы:
1. Какие вещества называют электролитами?
2. Почему электролиты называют проводниками электрического тока II рода? Какие вещества являются проводниками I рода?
3. Что называют степенью диссоциации электролита? Чему равна степень диссоциации сильного электролита?
4. Запишите уравнение электролитической диссоциации угольной кислоты и карбоната натрия. Объясните, в каком случае диссоциация протекает: а) обратимо; б) ступенчато.
5. Имеются сантимолярные растворы соляной и уксусной кислот. Объясните, в каком из них разница между значениями с Н+ и а Н более заметна.
6. Запишите три математические формулы, на основании которых можно: а) дать определение степени диссоциации электролита; б) вычислить степень диссоциации слабого электролита в растворе заданной концентрации; в) вычислить степень диссоциации слабого электролита, измерив электрическую проводимость его раствора.
7. Вычислите степень диссоциации NH3*H2O в его деци-, санти- и миллимолярных растворах. Постройте график зависимости степени диссоциации NH3*H2O от концентрации раствора. Ответ: 0,0134; 0,0423; 0,1338.
8. Что называют разведением раствора? Сделайте необходимые расчеты и постройте кривую зависимости степени диссоциации азотистой кислоты от разведения.
9. Вычислите концентрацию ионов Н+ в 0,1 М растворе СНзСООН и молярную электрическую проводимость этого раствора при 298 К, если при этой температуре μ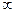 = 390,7 см2*Ом-1*моль-1. Ответ: 1,32*10-3 моль/л;
= 390,7 см2*Ом-1*моль-1. Ответ: 1,32*10-3 моль/л;
5,16 см2 *Ом-1*моль-1.
10. Почему при разбавлении раствора слабого электролита его удельная электрическая проводимость достигает максимума, а затем уменьшается, а молярная — достигает максимума μ,, а затем не изменяется (и в том, и в другом случаях Т = const).
11. Сопротивление ячейки, заполненной раствором КС1 с удельной электрической проводимостью 5,79*10-3 Ом-1*см-1, равно 103,6 Ом. Сопротивление той же ячейки, заполненной 0,01 М раствором уксусной кислоты, равно 5771 Ом. Определите молярную электрическую проводимость 0,01 М раствора уксусной кислоты. Ответ: 10, 39 см2*Ом-1*моль-1.
|
|
|
|
|
Дата добавления: 2013-12-12; Просмотров: 2491; Нарушение авторских прав?; Мы поможем в написании вашей работы!