КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Сульфування фенолу
|
|
|
|
Бромування фенолу
Фенол енергійно вступає в реакцію з галогенами. Він навіть вступає в реакцію з бромною водою, в результаті якої утворюється важкорозчинний у воді 2,4,6-трибромфенол. Ця реакція є якісною на феноли.
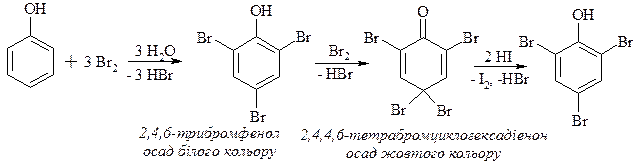
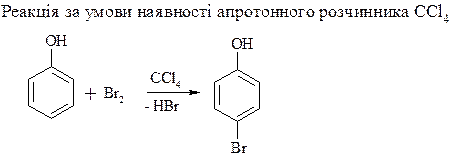

3. Нітрування фенолу
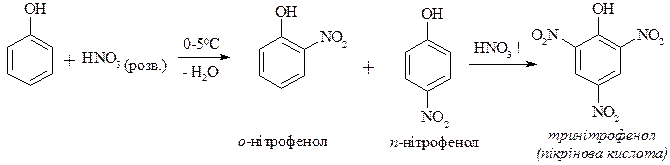
4. Нітрозування фенолу (реакція можлива тільки за рахунок донорних властивостей гідроксильної групи)
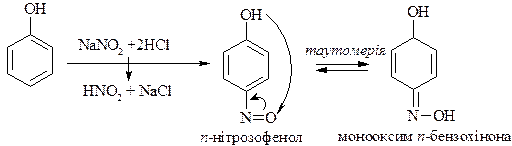
5. С-алкілування та С-ацилювання фенолу
· Реакції С-алкілування для фенолів проходять з малою швидкістю та малим виходом.
· Реакції С-ацилювання для фенолів проходять з малою швидкістю тому застосовують метод Фріза.
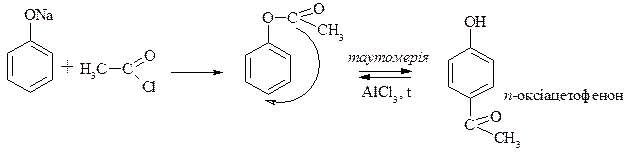
6. Гідроксиметилування фенолу
Феноли можуть вступати в реакції конденсації з багатьма речовинами, зокрема з альдегідами. Особливе значення має конденсація фенолу з формальдегідом, у результаті якої утворюється фенолформальдегідна смола. Ця реакція відбувається при наявності кислот або лугів. Низькомолекулярні полімерні сполуки називають новолаками:
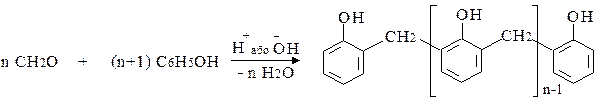
При нагріванні фенолу з надлишком формальдегіду утворюється полімер з розгалуженою просторовою сітчастою структурою – резит.
7. Рекція Кольбе-Шмідта – одержання саліцилової кислоти (аспірин)
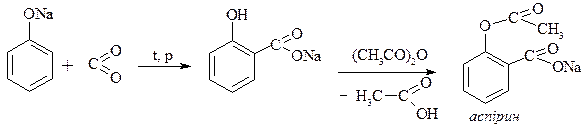
Феноли легко окиснюються, навіть киснем повітря. При окисленні хромовою сумішшю фенол перетворюється в пара-хінон 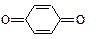 , який забарвлений у жовтий колір.
, який забарвлений у жовтий колір.
Двоатомні феноли:
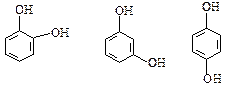
пірокатехін резорцин гідрохінон
а також три ізомери триатомних фенолів:
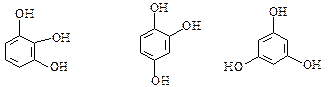 ,
,
пірогалол оксигідрохінон флороглюцин
які мають властивості близькі до властивостей одноатомних фенолів: виявляють кислотні властивості, утворюють ефіри та інші похідні за –ОН -групою, дають кольорові реакції з FeCl3, але легше окиснюються до відповідних хінонів.
|
|
|
Резорцин і флороглюцин мають здатність до кето-єнольної таутомерії:
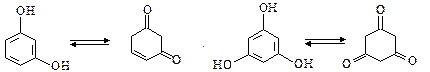
Внаслідок чого з оцтовим ангідридом вони реагують в єнольній формі, а з гідроксил аміном – в кетонній формі.
Хінони існують у вигляді двох ізомерів − о- і п-хінони:
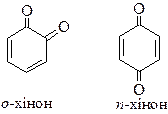
За структурою хінони – ненасичені циклічні кетони, які легко утворюються з двоатомних фенолів і легко відновлюються до двоатомних фенолів. Хінони вступають у реакції характерні для кетонів. Крім того хінони вступають у реакції 1,4- і 1,6-приєднання, які приводять до утворення похідних двоатомних фенолів. При змішуванні рівномолярних кількостей хінону і гідрохінону утворюється молекулярна сполука – хінгідрон.
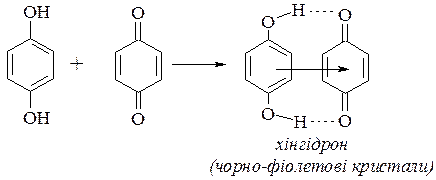
АЛЬДЕГІДИ І КЕТОНИ
Альдегіди і кетони – це сполуки, у молекулах яких міститься функціональна група 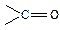 (карбонільна або оксогрупа). В альдегідах карбонільна група сполучена з вуглецевим радикалом і одним атомом водню
(карбонільна або оксогрупа). В альдегідах карбонільна група сполучена з вуглецевим радикалом і одним атомом водню 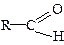 , у кетонах вона сполучена з двома вуглецевими радикалами:
, у кетонах вона сполучена з двома вуглецевими радикалами: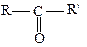
СПОСОБИ ОДЕРЖАННЯ АЛЬДЕГІДІВ І КЕТОНІВ
1. Реaкції окиснення:
a. Ненасичених вуглеводнів 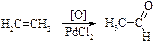
b. Насичених вуглеводнів
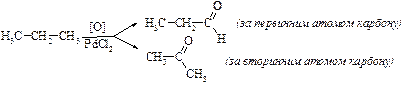
c. Ароматичних вуглеводнів
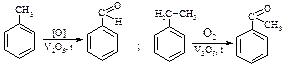
d. Спиртів
i. Первинні спирти
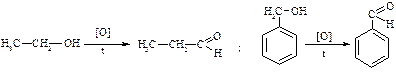
ii. Вторинні спирти
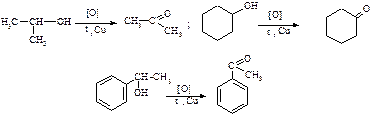
2. Каталiтичне дегідрування спиртів
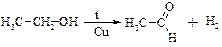
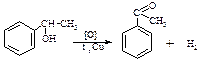
3. Гідроліз гемінальних дигалогеналканів
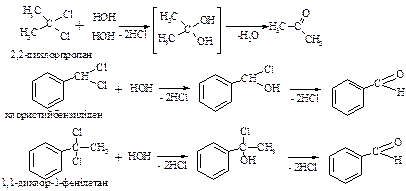
4. Декарбоксилювання (піроліз) солей карбонових кислот
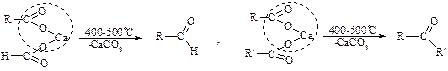
5. Гідратація алкінів за реакцією Кучерова
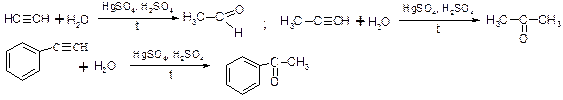
ОСОБЛИВОСТІ РЕАКЦІЙ НУКЛЕОФІЛЬНОГО ПРИЄДНАННЯ ДО АЛЬДЕГІДІВ І КЕТОНІВ
а) Електронні фактори
Пов'язані з тим, що алкільна група, як донор електронів подає електронну густину на карбонільний вуглець, що знижує частково позитивний заряд (δ+) на ньому.
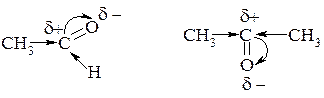
У випадку альдегідів таке зниження δ+ проходить менше, так як тільки одна алкільна група. Для кетону таке зниження δ+ на карбонільному атому карбону проходить більше, так як в цьму випадку має місце вплив двох алкільних груп.
б) Просторові фактори
|
|
|
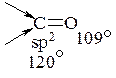
При приєднанні нуклеофілу до карбонільної групи атом карбону переходить з sp2-гібридного стану до sp3 –гібридного стану. Валентні кути змінюються від 1200 до 109028'. Тобто проходить зближення радикалів зв'язаних з карбонільною групою.
Для альдегідів таке зближення проходить легко, так як один з радикалів – це гідроген. Для кетонів таке зближення проходить трудніше і при наявності особливо об'ємних замісників (наприклад, трет -бутильних) реакційна здатність кетонів знижується настільки, що реакції нуклеофільного приєднання стають неможливими.
ВИСНОВОК: Альдегіди є більш реакційно-здатними сполуками ніж кетони.
Здатність альдегідів і кетонів вступати в реакції приєднання, приєднання-відщепленні визначається величиною частково позитивного заряду на атомі карбону карбонільної групи – чим більше δ+ на карбоні тим більшу реакційну здатність має альдегиди та кетони у вище зазначених реакціях.
Тому, реакційна здатність карбонільних сполук зменшується в такому ряду:
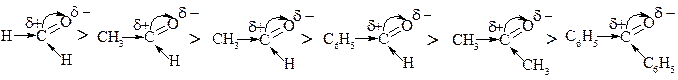
В цілому: а) альдегіди вступають в реакцію легше кетонів;
б) аліфатичні сполуки вступають в реакції легше алкілароматичних і тим більше чисто ароматичних.
Ароматичні альдегіди і кетони, які містять в орто- і особливо в пара-положеннях до карбонільної групи електоно-акцепторні замісники мають більш високу реакційну здатність за рахунок збільшення частково позитивного заряду (δ+) на атомі карбону карбонільної групи і навпаки: введення електроно-донорних замісників в тіж положення зменшують реакційну здатність альдегідів і кетонів за рахунок зниження заряду (δ+) на атомі карбону карбонільної групи.
Головна відзнака альдегідів від кетонів – альдегіди окиснюються дуже легко, особливо ароматичні, а кетони – важко.
Для альдегідів і кетонів характерні реакції нуклеофільного приєднання води, спиртів, синильної кислоти, гідросульфіту натрію, реактиву Гріньяра; реакції з азотовмісними сполуками (аміаком, гідроксиламіном, гідразином тощо); реакції конденсації (альдольна, кротонова), окиснення.
Механізм реакції нуклеофільного приєднання можна розглянути на прикладі реакції з HCN при лужному каталізі:
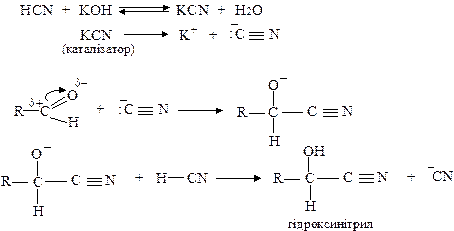
Реакція альдольної конденсації проходить в лужному середовищі і характерна лише для тих альдегідів і кетонів, що мають атом водню біля α-вуглецевого атому:
|
|
|
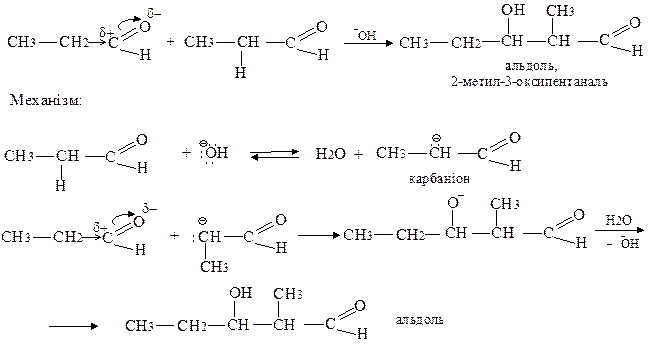
При нагріванні альдоль відщеплює воду з утворенням ненасиченого альдегіду. Ця реакція має назву кротонової конденсації.
Реакція альдольної конденсації характерна лише для тих альдегідів і кетонів, які мають рухливий атом водню біля a-вуглецевого атома відносно карбонільної групи.
Реакції полімеризації характерні лише для альдегідів. Особливе значення вони мають для формальдегіду. В водному розчині відбувається ступінчата полімеризація – утворюється параформ (параформальдегід):
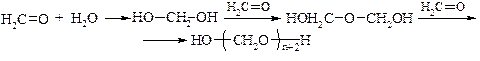
При нагріванні з параформу добувають формальдегід в газоподібному стані високої чистоти. В присутністі Н2SO4 відбувається циклічна полімеризація формальдегіду:
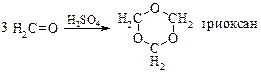
Перегрупування Бекмана. Тільки для ароматичних кетонів. Ароматичні кетони за своїми властивостями подібні до аліфатичних, вони дають тіж самі похідні, але чисто ароматичні не взаємодіють з бісульфітом натрію.
Із похідних ароматичних кетонів найбільше практичне значення мають оксими
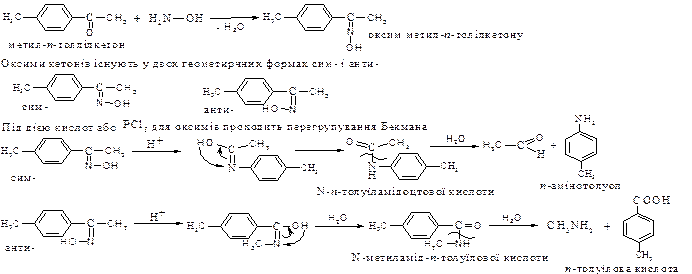 Значення перегрупування Бекмана. На основі ароматичних кетонів можна одержати різні аміни та кислоті: два ізомерних оксима утворюють два ізомерних аміда. Застосовується перегрупування Бекмана у виробництві синтетичного волокна – капрону.
Значення перегрупування Бекмана. На основі ароматичних кетонів можна одержати різні аміни та кислоті: два ізомерних оксима утворюють два ізомерних аміда. Застосовується перегрупування Бекмана у виробництві синтетичного волокна – капрону.
При окисненні альдегідів утворюються кислоти з тією ж кількістю атомів вуглецю. При окисненні кетонів утворюється суміш кислот. Для кетонів дія окисника спрямована на менш гідрогенізований атом вуглецю в α-положенні до карбонільної групи:
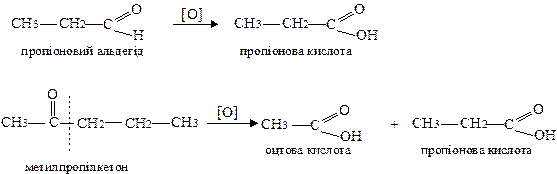
Реакції, що характерні тільки для альдегідів: окиснення аміачним розчином оксиду срібла (реакція срібного дзеркала) і полімеризація. За допомогою цих реакцій можна відрізнити альдегіди від кетонів.
АЗОТОВМІСНІ ОРГАНІЧНІ СПОЛУКИ
НІТРОСПОЛУКИ
Похідні вуглеводнів в молекулах яких є група –NO2.
Гомологічний ряд: R–NO2 CnH2n+1NO2.
КЛАСИФІКАЦІЯ
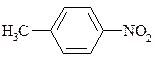
| АЛІФАТИЧНІ | ЖИРНО-АРОМАТИЧНІ | АРОМАТИЧНІ |
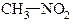
| 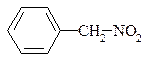
| 
|
| нітрометан | α -нітротолуол, фенілнітрометан | п -нітротолуол |
|
|
|
ІЗОМЕРІЯ І НОМЕНКЛАТУРА
В залежності від характеру атома карбону з яким зв’язана нітрогрупа аліфатичні нітросполуки бувають:
| первинні | вторинні | третинні |
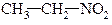
| 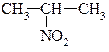
| 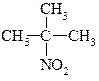
|
| нітроетан | 2-нітропропан | 2-нітро-2-метилпропан |
Для нітросполук характерна структурна ізомерія, що обумовлена розгалуженням вуглецевого ланцюга та ізомерія положення нітрогрупи.
СПОСОБИ ОДЕРЖАННЯ НІТРОСПОЛУК
Одержання аліфатичних нітросполук
1. Рідкофазне нітрування алканів за Коноваловим
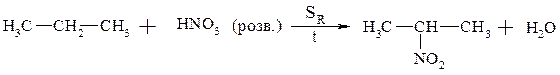
2. Нуклеофільне заміщення галогену на нітрогрупу – алкілування галоїдними алкілами солей азотистої кислоти
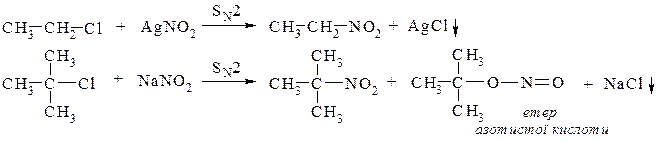
3. У промислвості проводять парофазне нітрування алканів t = 300-4000C, концентрована сірчана кислота, але таке нітрування супроводжується деструкцією алканів (окисненя і крекінг).
Способи одержання жирно-ароматичних нітросполук
1. Рідкофазне нітруваня за Коноваловим
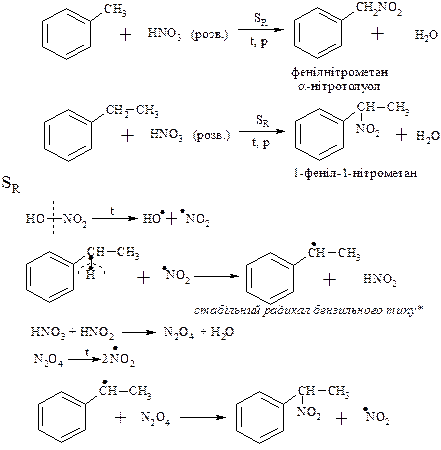
При нітруванні бокового ланцюга з двома і більше атомами карбону легше всього реакція проходить за a-вуглецевим атомом бокового ланцюга у звязку із більшою стабілізацією радикалу бензільного типу* (цей радикал стабільний за рахунок супряження неспаренного електрону атома карбону бокового ланцюга з p-електронним секстетом ароматичного ядра).
2. Нуклеофільне заміщення галогену на нітрогрупу
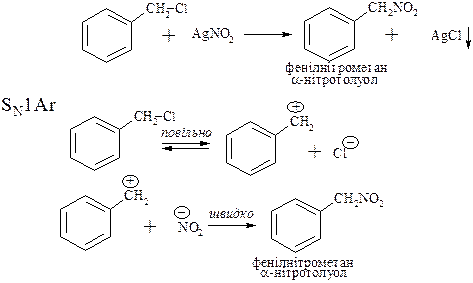
Одержання ароматичних нітросполук
Одержуються реакцією електрофільного заміщення – нітруванням.
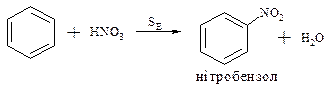
1. Як нітруючий засіб використана азотна кислота, яка диссоциює у водному розчині, але концентрація +NO2 у даному випадку дуже мала із-за зворотньої ракції.
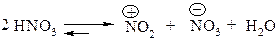
2. Ароматичні сполука частіше всього нітрують нітруючою сумішшю. Роль сірчаної кислоти зводиться до збільшення в рівноважній суміші концентрації +NO2 за семою:
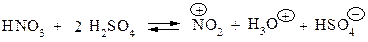
3. Гомологи бензолу нітруються легше ніж сам бензол. Для одержання ди- і три- ароматичних нітропохідних нітрування проводять в більш жорстких умовах.
ХІМІЧНІ ВЛАСТИВОСТІ
БУДОВА НІТРОГРУПИ
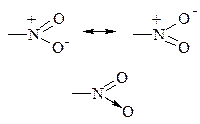
| Атом нітрогену має п’ять валентних електронів: один електрон витрачає на утворення ковалентного зв’язку з любим радикалом; два електрони витрачає на утворення ковалентного полярного зв’язку з одним із атомів оксигену і два електрони витрачає на утворення донорно-акцепторного (семіполярного) зв’язку з іншим атомом оксигену. |
Нітрогрупа є одним із найбільш сильних акцепторів електронів, так як на атомі нітрогену локалізований цілий позитивний заряд, що з одного боку приводить до різкого збільшення рухливості протону у α-вуглецевого атому (більш сильному ніж цей ефект у альдегідах і кетонах), а з іншого боку до дезактивації ароматичного кільця до реакцій електрофільного заміщення і полегшення реакцій нуклеофільного заміщення.
І. РЕАКЦІЇ ЗА НІТРОГРУПОЮ
1. Реакції відновлення з утворенням амінів
a. Для аліфатичних нітросполук
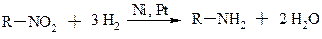
b. Для ароматичних нітросполук
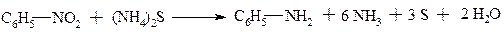
В якості відновників використовують: залізо в оцтовій кислоті: залізо, цинк у соляній кислоті, молекулярний гідроген у присутності нікеля, платини, тощо.
Відновлення ароматичних нітросполук проходить у нейтральному, слабо кислому та лужному середовищах. Відрізняють різними механізмами.
Механізм відновлення у нейтральному і слабокислому середовищах.
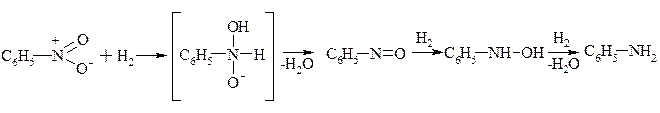 Механізм відновлення у лужному середовищі.
Механізм відновлення у лужному середовищі.
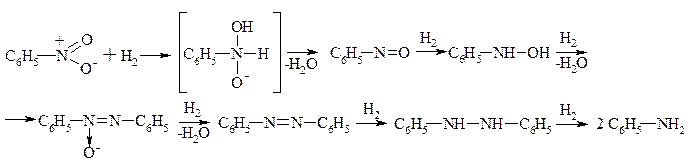 2. Ацинітротаутомерія
2. Ацинітротаутомерія
Первинні і вторинні нітросполуки, які містять рухливий атом гідрогену в α-положенні по відношенню до нітрогрупи розчиняються у лугах з утворенням солей – це явище ацинітротаутомерія.
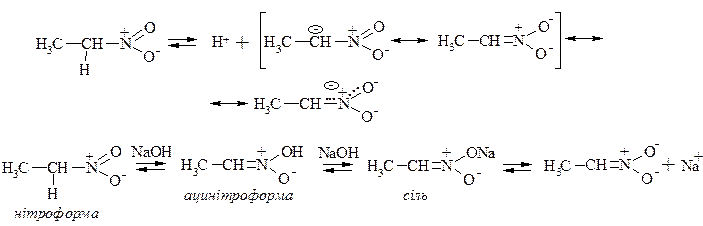 Ацинітроформа є більш сильною кислотою ніж нітроформа. Ці форми відрізняються місцем розташування протону – таутомери.
Ацинітроформа є більш сильною кислотою ніж нітроформа. Ці форми відрізняються місцем розташування протону – таутомери.
Таутомерія – існування у рухливій рівновазі двох різних форм.
3. Реакції, які відрізняють нітросполуки з нітрогрупою у боковому ланцюзі від ароматичних нітросполук з нітрогрупою у ядрі.
Для нітросполук, які містять нітрогрупу у боковому ланцюзі характерне явище ацинітротаутомерії, так як в цьому випадку є рухливий атом гідрогену.
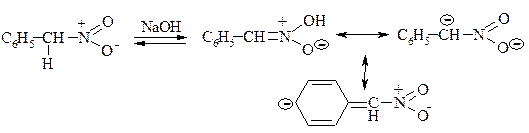
4. Взаємодія з азотистою кислотою – реакція нітрозування.
Реакція дозволяє розрізнити первинні, вторинні і третинні нітросполуки.
1. Первинні нітросполуки.
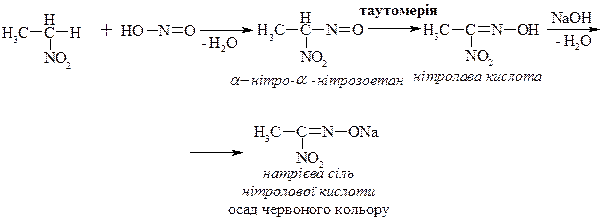
2. Вторинні нітросполуки.

3. Третинні нітросполуки. Ці сполуки не мають рухливого атома гідрогену, тому в реакцію нітрозування не вступають.
5. Взаємодія нітросполук з альдегідами – реакція конденсації.
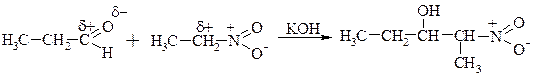
ІІ. РЕАКЦІЇ ЗА АРОМАТИЧНИМ ЯДРОМ.
1. Реакції електрофільного заміщення. Нітрогрупа як акцептор електронів зменшує електронну густину в бензольному ядрі і реакції електрофільного заміщення утруднює, в результаті утворюються м -ізомери.
2. Реакції нуклеофільного заміщення. За тими ж причинами нітргрупа реакції нуклеофільного заміщення полегшує, в результаті утворюються о - і п -ізомери.
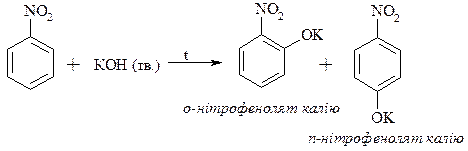
3. Акцепторні властивості нітрогрупи. У випадку о - і п -нітрохлорбензолів під впливом нітрогрупи галоген має високу рухливість і легко заміщується на інші нуклеофіли.
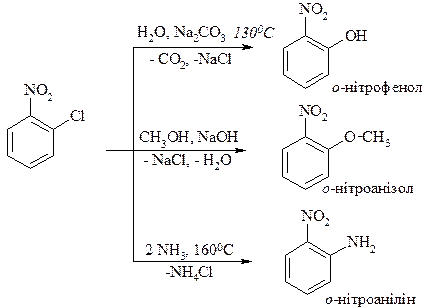
АМІНИ
Аміни – похідні аміаку, в молекулі якого один або декілька атомів водню заміщені на вуглеводневий радикал (як аліфатичний, так і ароматичний).
КЛАСИФІКАЦІЯ
| АЛІФАТИЧНІ | ЖИРНО-АРОМАТИЧНІ | АРОМАТИЧНІ | |
| ПЕРВИННІ | 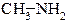 метиламін
метиламін
| 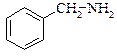 бензиламін
бензиламін
| 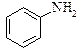 анілін
анілін
|
| ВТОРИННІ | 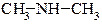 диметиламін
диметиламін
| 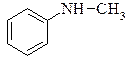 N-метиланілін
N-метиланілін
| 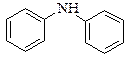 дифеніламін
дифеніламін
|
| ТРЕТИННИ | 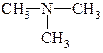 триметиламін
триметиламін
| 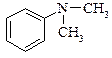 N,N-диметиланілін
N,N-диметиланілін
| 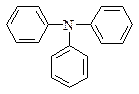 трифеніламін
трифеніламін
|
СПОСОБИ ОДЕРЖАННЯ АМІНІВ
ДЛЯ ПЕРВИННИХ АМІНІВ
1. Відновлення нітросполук за Зініним
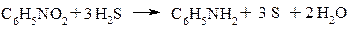
2. Відновлення азотовмісних сполук
a. Відновлення оксидів
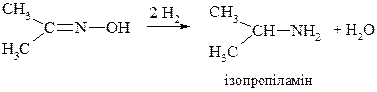
b. Відновлення нітрилів
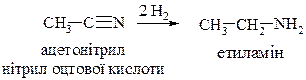
c. Відновлення амідів карбонових кислот
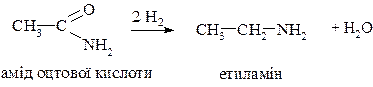
3. Алкілування
a. Алкілування аміаку галоїдними алкілами
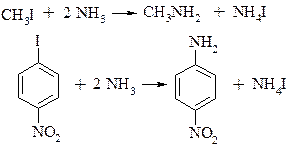
b. Алкілування аміаку спиртами
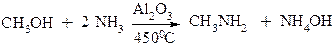
Реакції а) і b) приводять до утворення суміші первинних, вторинних і третинних амінів, а також до четвертинних амонійних солей.
4. Реакція Гофмана
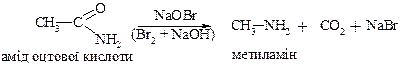
ДЛЯ ВТОРИННИХ АМІНІВ
1. Відновлення ізонітрилів
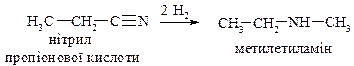
За цією реакцією одержують вторинні аміни, які обов’язково містять метильну групу з одного боку.
2. Метод допоміжного ацилювання (для жирно-ароматичних амінів)
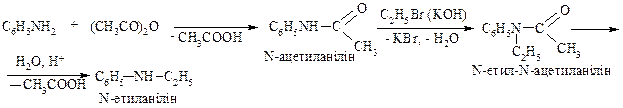
3. Алкілування амінів
a. Галоїдними алкілами
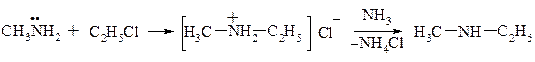
b. Спиртами
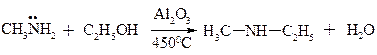
Реакції а,b також приводять до утворення суміші вторинних і третинних амінів.
ДЛЯ ТРЕТИННИХ АМІНІВ
1. Алкілування амінів галоїдними алкілами або спиртами
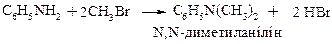
2. Арилювання вторинних амінів
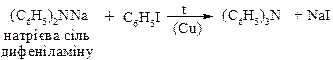
ХІМІЧНІ ВЛАСТИВОСТІ
1. Основність амінів
Основні властивості амінів визначаються наявністю на атомі азоту пари електронів і її здатністю приєднувати протон. Аміни як правило є більш сильними основами ніж вода, так як азот менш електронегативний елемент ніж кисень і відповідно легше віддає свою пару електронів для протонування.
Основність амінів в значній мірі залежить від їх будови. Аліфатичні аміни є більш сильними основами ніж ароматичні.
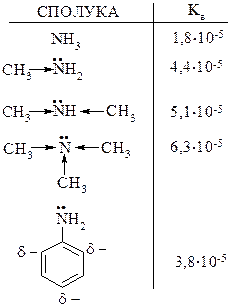
Порівняння основності амінів
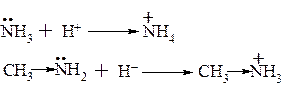
Катіон амонію менш стійкий ніж катіон метиламонію. У зв’язку з позитивним індукційними ефектом метильної групи позитивний заряд на атомі азоту частково погашається, тобто стійкість даного катіону збільшується. У аліфатичному ряді в протонних розчинниках вторинні аміни більш сильні основи ніж первинні, а третинні аміни мають менші основні властивості ніж вторинні, що пояснюється гіршою сольватацією розчинників із-за просторових факторів.
Ароматичні аміни мають нижчі основні властивості ніж аліфатичні і аміак, за рахунок супряження неподільної пари електронів атома азоту з p-електронами бензольного кільця, що значно зменшує електронну густину на атомі азоту, тобто зменшує його нуклеофільність.
Атом азоту аміно групи, яка зв’язана з бензольним кільцем, знаходиться в стані близькому до sp2-гібридизованого стану в результаті супряження знижується здатність неподільної пари електронів азоту приєднувати протон.

Крім того, при утворенні солей порушується супряження аміно групи з ароматичним ядром (азот переходить в sp3-гібридизований стан), що енергетично не вигідно:
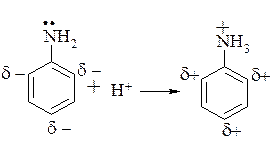
АРОМАТИЧНІ ДІАЗО- І АЗОСПОЛУКИ
Діазосполуки Ar-N2X це органічні сполуки з функціональною групою, що складається із двох атомів азоту, які сполучені з ароматичним радикалом і неорганічним замісником Х.
Для ароматичних діазосполук характерна наявність діазогрупи, що сполучена з вуглеводневим радикалом.
Ароматичні діазосполуки існують у вигляді декількох малостійких форм, які легко переходять одна в одну. До них відносяться:
1. Солі діазонію 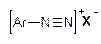 , де Х¯ – аніон сильної кислоти (HSO4¯, Cl¯ та інші).
, де Х¯ – аніон сильної кислоти (HSO4¯, Cl¯ та інші).
2. Діазогідрати 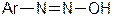 , які мають амфотерні властивості: у кислому середовищі вони утворюють діазокатіони
, які мають амфотерні властивості: у кислому середовищі вони утворюють діазокатіони 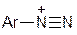 , а в лужному – діазотат - аніони
, а в лужному – діазотат - аніони 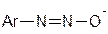 .
.
3. Діазосполуки 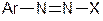 , де Х – залишок слабкої кислоти. Такі сполуки не відносяться до солей діазонію, не мають іонних зв’язків і не дисоціюють. До них належать арилдіазосульфіти
, де Х – залишок слабкої кислоти. Такі сполуки не відносяться до солей діазонію, не мають іонних зв’язків і не дисоціюють. До них належать арилдіазосульфіти 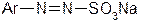 , арилдіазоціаніди
, арилдіазоціаніди 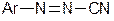 та інші.
та інші.
Із всіх діазосполук найбільше значення мають солі арилдіазонію. Одержують їх реакцією діазотування – дією на водний розчин первинного ароматичного аміну нітритом натрію у присутності надлишку мінеральної кислоти при охолодженні:

Будова діазогрупи арилдіазонію є проміжною між двома граничними структурами
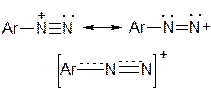
Солі арилдіазонію у водному розчині змінюють свою будову в залежності від значення рН середовища: при нейтралізації кислого середовища вони переходять у форму діазогідрата, який у лужному середовищі утворює діазотат–аніона:
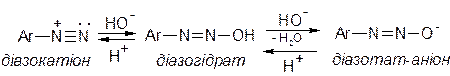
Для діазосполук характерні два типи хімічних перетворень – реакції з виділенням азоту і реакції без виділення азоту. Перший тип хімічних перетворень має велике значення для синтезу різноманітних сполук, другий – є основою цілої галузі промисловості – одержання азобарвників.
1. Реакції діазосполук з виділенням азоту
Такі реакції використовуються для одержання різних похідних бензолу, у тому числі маталоорганічних сполук, а також для вилучення (елімінування) аміногрупи. Механізм таких реакцій SN1 i SR.
А. Гетеролітичний розклад солей діазонію. Такими реакціями без каталізатора можна замінити діазогрупу на гідрокси-групу (–OH), метокси-групу (–OCH3), фтор (–F).
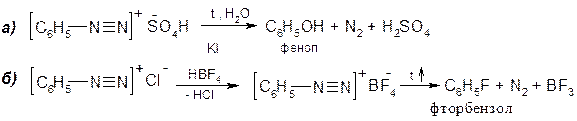
Розкладання гідросульфату фенілдіазонію водою відбувається за механізмом SN1:
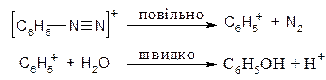
Б. Радикальний некаталітичний розклад солей діазонію. За цим механізмом відбувається заміщення діазогрупи на йод. Загальна схема реакції:
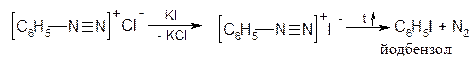
Механізм реакції:
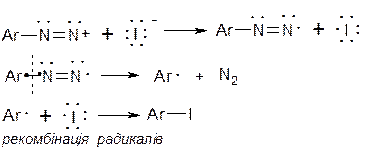
В. Радикальний каталітичний розклад солей діазонію. Такими реакціями діазогрупу можна замінити на Cl, Br, -CN, -SCN, -NO2. Реакції проходять при наявності каталізатору – відповідної солі одновалентної міді. Це реакція Зандмейєра.
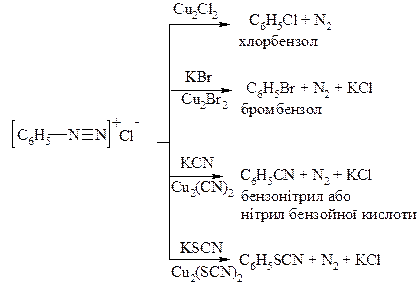
Механізм – радикальний
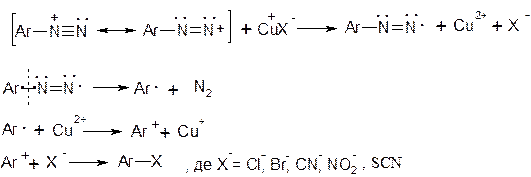
Механізм заміщення діазогрупи на водень:
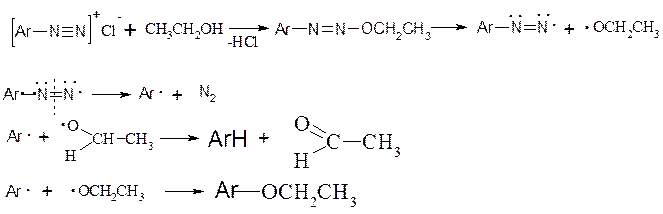
2. Реакції без виділення азоту
Реакції азосполучення, що приводять до утворення барвників. Азосполучення – це взаємодія солей арилдіазонію з похідними ароматичних вуглеводнів (амінів, фенолів). Азосполучення приводить до утворення азосполук, в молекулах яких присутня азогрупа –N=N–, яка знаходиться в спряженні з ароматичною π-електронною системою. Оскільки діазокатіон достатньо слабкий електрофіл, в реакцію азосполучення вступають вуглеводні, що мають сильні електронодонорі групи, наприклад, –NH2, -OH. Середовище реакційної маси при цьому не повинно бути дуже кислим, так як аміногрупа –NH2 може перейти амонійну –N+H3, яка є акцептором електронів, і не дуже лужним, так як діазокатіон перейде у форму діазотатаніону, який не вступає в реакцію азосполучення, так як це не електрофіл, а нуклеофіл.
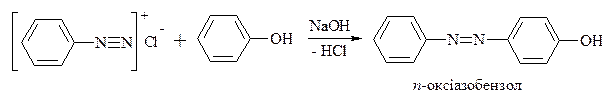
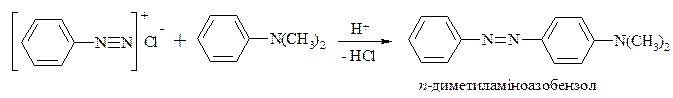 Механізм:
Механізм:
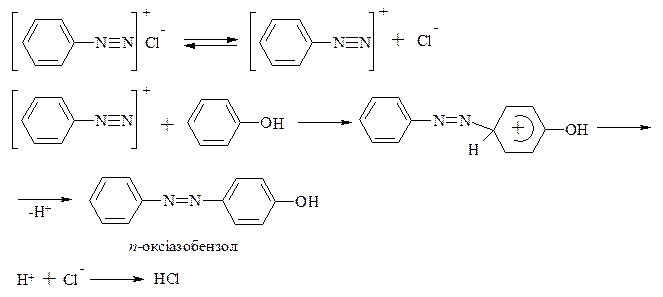
Азобарвники застосовують для забарвлення самих різних матеріалів, в першу чергу, волокон (рослинних, тваринних та штучних) і тканин; також пластмас, гуми, деревини, шкіри і харчових продуктів. Вони використовуються для виготовлення лаків, покривних і поліграфічних барвників, а також як індикатори.
Важлива властивість солей діазонію – це їх здатність вступати в реакції відновлення. Ця властивість солей діазонію використовується при встановленні будови азобарвника.
Так при дії лужного розчину гідросульфіту натрію азобарвник легко розщеплюється на два аміни, що дає можливість встановити будову вихідного барвника.
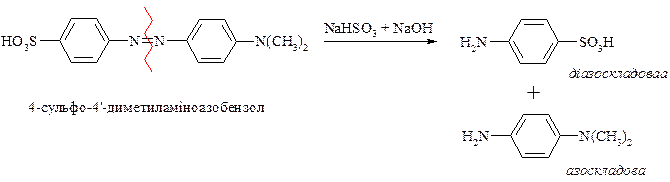
КАРБОНОВІ КИСЛОТИ
Карбонові кислоти – вуглеводні, які в своєму складі мають карбоксильну групу COOH.
Загальна формула
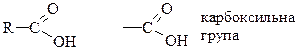
За числом карбоксилів – одно- та багатоосновні.
За природою вуглеводневого радикалу R бувають – насичені, ненасичені, аліфатичні та циклічні, ароматичні і т.д.
Номенклатура ІЮПАК дозволяє використання тривіальних назв.
| Структура | Трев назва | ІЮПАК | рКа |
| HCOOH | Мурашина | Метанова | 3,75 |
| CH3COOH | Оцтова | Етанова | 4,76 |
| C2H5COOH | Пропіонова | Пропанова | 4,89 |
| C3H7COOH | Масляна | Бутанова | 4,82 |
| (CH3)2CHCOOH | Ізомасляна | Ізобутанова | |
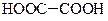
| Щавелева | Етандіонова | 1,27 |
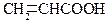
| Акрілова | Пропенова | 4,26 |
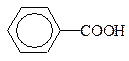
| Олеїнова,бензойна | Бензолкарбонова | |
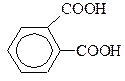
| фталева | 1,2-бензолдікарбонова | 2,95 |
|
|
|
|
|
Дата добавления: 2014-01-05; Просмотров: 7589; Нарушение авторских прав?; Мы поможем в написании вашей работы!