КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Основні залежності і розрахункові формули 2 страница
|
|
|
|
3.6.1. Екстрактивна ректифікація
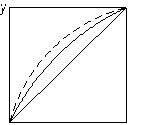
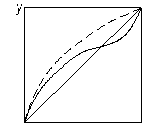
На рис. 3.25 показано вплив додавання розділювального компонента на зміну відносної леткості компонентів бінарної суміші. Пунктиром зображено криві рівноваги, одержані під час розділення суміші близькокиплячих компонентів (рис. 3.25 а) і азеотропної суміші (рис. 3.25 б) у присутності третього компонента. З діаграми у – х видно, що внаслідок різкого підвищення відносної леткості процес розділення значно полегшується, його можна здійснити за меншої кількості ступенів розділення.
а |
б |
Рис. 3.25. Вплив добавки розділювального компонента під час екстрактивної ректифікації: а – суміш близькокиплячих компонентів; б – азеотропна суміш
У схемі установки для екстрактивної ректифікації (рис. 3.26) початкову суміш, що складається з компонентів А+В, подають на живлячу тарілку екстрактивно-ректифікаційної колони 1, яка зрошується згори розділювальним агентом С, більш висококиплячим, ніж компоненти А і В. Компонент В добре розчинний в С, тоді як компоненти А і С взаємно нерозчинні або погано розчинні один в одному. У результаті компонент С екстрагує компонент В (більш висококиплячій у цій суміші) з рідкої та парової фази. Суміш В+С видаляється у вигляді залишку, а дистилят є чистим компонентом А.
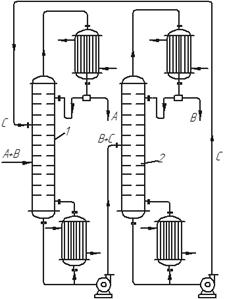
Рис. 3.26. Схема установки для екстрактивної ректифікації:
1 – колона екстрактивно-ректифікаційна; 2 – ректифікаційна колона
для регенерації розділювального агента
За процесом екстрактивної ректифікації йде процес розділення суміші компонентів В+С в ректифікаційні колоні 2. У цій колоні дистилятом є компонент В, більш леткий, ніж С. Регенерований розділювальний компонент С видаляється знизу колони 2 та надходить у колону 1 для повторного використання. Отже, додавання екстрагуючого компонента та розчинення в ньому компонента вихідної суміші ускладнює установку.
|
|
|
Типовим прикладом застосування екстрактивної ректифікації є розділення суміші близькокиплячих бензолу (компонент А) іциклогексану ( компонент В) із застосуванням фенолу (компонент С) як розділювального агента. Екстрактивна ректифікація здійснюється тільки безперервним способом.
3.6.2. Азеотропна ректифікація
Під час азеотропної ректифікації зазвичай використовують розділяючий компонент С, який утворює з одним з компонентів початкової суміші (А або В) азеотропну суміш, що має мінімальну температуру кипіння. Утворена більш летка, ніж початкова, азеотропна суміш відганяється як дистилят, а інший, практично чистий, компонент видаляється у вигляді залишку.
Іноді можна підібрати розділювальний агент, що створює з одним з компонентів початкової суміші нову азеотропну суміш з максимальною температурою кипіння. У цьому разі нова азеотропна суміш видаляється у вигляді залишку, а згори колони відбирають дистилят, чистий інший компонент початкової суміші. Можна також здійснити азеотропну ректифікацію за допомогою розділювального компонента, що створює азеотропні суміші з обома компонентами. При цьому співвідношення компонентів А і В у потрійній азеотропній суміші повинно бути іншим, ніж в початковій суміші, що надходить на розділення. За цим варіантом процесу дистилят, що видаляється з колони, є леткою азеотропною сумішшю (з трьох компонентів), а залишок – один з компонентів початкової суміші практично у чистому вигляді.
Підбір розділювального агента здійснюють на основі другого закону Вревського, що вказує напрям зміни складу азеотропної суміші з температурою.
В установці для азеотропної ректифікації (рис. 3.27), що здійснюється з утворенням азеотропної суміші, яка має мінімальну температуру кипіння, вихідна азеотропна суміш (А + В) надходить на живлячу тарілку колони 1, яка зрошується зверху розділювальним агентом С. Згори колони видаляється азеотропна суміш компонентів А + С з мінімальною температурою кипіння (дистилят), знизу колони виходить компонент В (залишок).
|
|
|
На рис. 3.27 показано варіант процесу азеотропної ректифікації, коли азеотропна суміш, що утворюється, складається з компонентів із різною взаємною розчинністю за різних температур.
У цьому разі компоненти А і С, що знаходяться у рідкому стані, є практично взаємно нерозчинні. Тому дистилят після охолоджування розділяється на компоненти А і С у відстійнику 2. Компонент А є кінцевим продуктом, а регенерований компонент С після нагрівання в підігрівачі 3 повертається на зрошування колони 1. У схемі, показаній на рис. 3.27, в дефлегматорі колони 1 конденсується тільки частина пари (А + С), що є необхідною для отримання флегми, а решта зріджується і охолоджується в холодильнику-конденсаторі перед надходженням у відстійник 2.
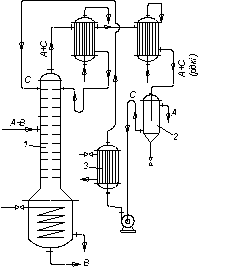
Рис. 3.27. Схема установки для азеотропної ректифікації:
1 – ректифікаційна колона; 2 – відстійник (сепаратор); 3 – підігрівач
Як приклад застосування азеотропної ректифікації можна вказати на процес розділення азеотропної суміші “етиловий спирт – вода” (температура кипіння
~ 78°С), де як розділювальний компонент використовують бензол, що створює з водою і спиртом потрійну азеотропну суміш з мінімальною температурою кипіння (~ 64,8°С). Залишок, що видаляється з колони, є безводним етиловим спиртом.
Процеси азеотропної ректифікації здійснюють безперервним і періодичним способами, причому в останньому випадку компонент, який розділяють, повністю завантажується в куб колони разом з початковою сумішшю, що спрощує схему установки.
Під час азеотропної ректифікації зазвичай є необхідною більша витрата тепла, ніж під час екстрактивної ректифікації. Крім цього, під час азеотропної ректифікації складніше підібрати розділювальний агент і обмеженою є можливість зміни співвідношення його кількості і кількості вихідної суміші порівняно з екстрактивною ректифікацією.
|
|
|
Як розділювальний агент під час екстрактивної і азеотропної ректифікації більшого поширення набувають розчинні тверді речовини, зокрема солі, у присутності яких в необхідний бік змінюється співвідношення компонентів, що розділяються, за фазової рівноваги.
Рідини, що практично не змішуються і частково змішуються, в хімічній технології розділяють гетерогенною азеотропною ректифікацією (рис. 3.28). Наприклад, так вилучають органічні речовини від невеликих домішок розчиненої в них вологи. Процес здійснюють у вичерпних колонах 1 і 2. Початкова суміш, що складається з компонентів А і В, надходить у відстійник 3, де змішується з конденсатом з дефлегматора 4, який є загальним для обох колон. У відстійнику цей конденсат розшаровується на два шари, склади яких відповідають взаємній розчинності компонентів.
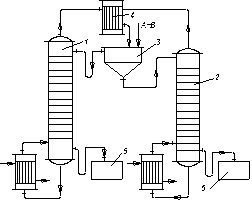
Рис. 3.28. Схема двоколонної установки для гетероазеотропної ректифікації
бінарних рідин, що розшаровуються: 1,2 – ректифікаційні колони;
3 – відстійник; 4 – дефлегматор; 5,6 – збірники
Розчин А і В (верхній шар) зливається в колону 1; тут в результаті перегонки утворюється пара, що є гетероазеотропом, який надходить з дефлегматора 4 у відстійник 3. Залишок з колони 1 є практично чистим компонентом В, який зливається в збірку 5.
Розчин В і А (нижній шар) розділяється в колоні 2 на гетероазеотроп і компонент А. Гетероазеотропблизький за складом до гетероазеотропу, що відгониться з колони 1, тому він прямує через загальний дефлегматор 4 у відстійник 3. Знизу колони 2 відводиться в збірник 6 практично вільний від домішок компонент А.
3.6.3. Молекулярна дистиляція
Вище було розглянуто плівкову ректифікацію під вакуумом, за допомогою якої розділяють нестійкі органічні сполуки. Проте температура кипіння багатьох високомолекулярних речовин (з молекулярною вагою ³300) навіть за значного вакууму залишається дуже високою для того, щоб їх можна було розділити, не побоюючись розкладання. Крім того, для багатьох сумішей необхідно звести до мінімуму тривалість розділення.
|
|
|
Такі суміші розділяють, створюючи дуже глибокий вакуум над поверхнею рідини, що відповідає залишковому тиску 10-3–10-4 мм рт. ст. В умовах глибокого вакууму із зменшенням густини газу зростає довжина вільного пробігу молекул і за достатньо низького залишкового тиску вона може стати більшою від відстані між поверхнями випаровування і конденсації. При цьому велика частина молекул, що відриваються з поверхні випаровування, потрапляє на поверхню конденсації і не повертається з цієї поверхні. Процес здійснюється за наявності близько розміщених поверхонь випаровування і конденсації.
Процес молекулярної дистиляції відбувається випаровуванням рідини з її поверхні за відсутності кипіння. Тому, на відміну від ректифікації, молекулярна дистиляція не характеризується деякими сталими температурою і тиском.
Під час молекулярної дистиляції молекули пари видаляються з поверхні випаровування зразу ж після їх утворення, і рівновага між парою і рідиною не встигає встановитися. Тому розділювальний ефект молекулярної дистиляції визначається не відношенням тиску насиченої пари компонентів суміші або відносною леткістю a, а відношенням швидкостей випаровування компонентів суміші, або коефіцієнтом розділення aм.
Швидкість випаровування 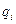 будь-якого компонента ідеального розчину є пропорційною до його мольної частки
будь-якого компонента ідеального розчину є пропорційною до його мольної частки 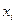 в рідині. Згідно з молекулярно-кінетичною теорією газів
в рідині. Згідно з молекулярно-кінетичною теорією газів
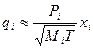 , (3.27)
, (3.27)
де 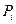 – тиск насиченої пари чистого компонента за температури кипіння суміші; Mi – молекулярна вага компонента; Т – абсолютна температура.
– тиск насиченої пари чистого компонента за температури кипіння суміші; Mi – молекулярна вага компонента; Т – абсолютна температура.
Отже, для бінарної суміші компонентів 1 і 2 можна записати:
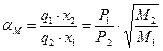 . (3.28)
. (3.28)
Згідно з цим виразом, ступінь розділення під час молекулярної дистиляції є більшою, ніж за рівноважною в 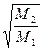 разів.
разів.
Процес молекулярної дистиляції складається з дифузії молекул переважно НК з глибини шару (плівки) рідини до поверхні випаровування, переміщення молекул пари на поверхню конденсації і їх конденсації на цій поверхні. В умовах звичайної дистиляції рідина інтенсивно перемішується під час кипіння з утворенням бульбашок, що піднімаються, і концентрації компонентів вирівнюються в об’ємі рідини. За молекулярної дистиляції швидкість випаровування компонента є пропорційною до його концентрації в рідині (за інших однакових умов). Тому зменшення концентрації компонента в рідині спричиняє зменшення швидкості його випаровування і погіршення розділення. Отже, ефективність процесу залежить від співвідношення швидкостей дифузії (в рідкій фазі) і випаровування компонента. Зазвичай дифузія компонента в рідині є повільнішим процесом, і молекулярну дистиляцію треба здійснювати в умовах, що сприяють прискоренню цієї лімітуючої стадії.
Як відомо з розділу I, швидкість дифузії в рідині можна збільшити за зростання швидкості руху і турбулізації шару рідини, а також за зменшення його товщини. Ці умови створюють в апаратах для молекулярної дистиляції (див. нижче).
Для оцінки ступеня розділення за молекулярної дистиляції використовують поняття про теоретичну молекулярну тарілку (ТМТ), що відповідає ступеню розділення, за якого співвідношення мольних концентрацій компонентів в дистиляті дорівнює відношенню швидкостей випаровування компонентів за повного перемішування шару дистильованої рідини. У виробничих умовах за разового випаровування ступінь розділення коливається від 0,3 до 0,95 ТМТ.
В установці для молекулярної дистиляції (рис. 3.29) на початку процесу необхідно видалити з вихідної суміші розчинені в ній гази і швидко відкачати їх з апарата. Початкова суміш зі сховища 1 надходить в багатоступінчатий дегазатор 2, де у разі нагрівання до 70–100 °С відбувається виділення з неї газів. Гази відкачуються форвакуум-насосами 3 (з перших ступенів) і спареними з ними дифузійно-конденсаційними насосами 4 (з останніх ступенів). Після цього суміш надходить або безпосередньо, або через підігрівач в дистиляційний апарат 5, з якого гази і пару відкачують спареними насосами 3 і 4. Дистилят і залишок прямуютьв окремі збірники 6. Між апаратом і насосами іноді встановлюють пастки 7, охолоджувані холодоагентом, щоб запобігти потраплянню в насоси пари води і органічних рідин.
Для молекулярної дистиляції застосовують плівкові апарати різних конструкцій, описані в спеціальній літературі.
На рис. 3.30 показано промисловий одноступінчастий відцентровий апарат (з плівкою, що піднімається) для молекулярної дистиляції. В корпусі 1 обертається алюмінієвий ротор-випарник 2 конічної форми, що обігрівається зовні електричним нагрівачем 3. Швидкість обертання ротора становить приблизно
400 хв -1. Усередині ротора знаходиться охолоджуваний зсередини гарячої водою конденсатор 5, виготовлений у вигляді розміщених віялоподібних плоских порожнистих елементів. Відстань між внутрішньою поверхнею ротора 2 і поверхнею конденсатора 5 становить 20–30 мм.
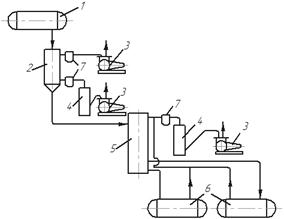
Рис. 3.29. Схема установки для молекулярної дистиляції: 1 – сховище початкової суміші; 2 – дегазатор; 3 – форвакуум-насоси; 4 – дифузійно-конденсаційні насоси;
5 – дистиляційний апарат; 6 – збірники; 7 – пастки
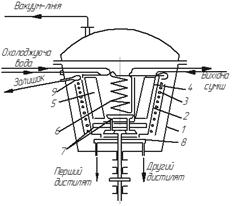
Рис. 3.30. Одноступінчастий відцентровий апарат (з плівкою, що піднімається)
для молекулярної дистиляції: 1 – корпус; 2 – ротор-випарник; 3 – електричний нагрівач; 4 – труба для подавання вихідної суміші; 5 – конденсатор;
6 – охолоджуваний змійовик; 7 – піддон для збору дистиляту; 8 – кільцевий жолоб
для відведення дистиляту; 9 – жолоб для відведення залишку
Рідина, що зазвичай є багатокомпонентною сумішшю, трубою 4 надходить на дно ротора. Під дією відцентрової сили ця рідина у вигляді тонкої плівки турбулентно піднімається вгору нагрітою поверхнею ротора і випаровується у міру піднімання. Пара менш летких компонентів конденсується на поверхні конденсатора 5, а пара більш летких – на поверхні конденсатора-змійовика 6, що охолоджується холодною водою. Рідина стікає в піддони 7, встановлені під конденсаторами, а з них – в кільцеві жолоби 8, звідки трубками видаляються роздільно дві фракції дистиляту. Залишок переливається через верхній край ротора в жолоб 9 і відводиться з апарата.
Молекулярна дистиляція є дорогим способом розділення. Її застосовують у виробництвах деяких пластмас, вітамінів, масел і мастил, жирних кислот, ефірів і ін.
3.6.4. Низькотемпературна ректифікація
Розділення зріджених газових сумішей ректифікацією здійснюють за дуже низьких температур під надлишковим тиском в апаратах, дещо відмінних від звичайних. При цьому продукти розділення одержують повністю або частково у вигляді пари. Проте основні закономірності процесу розділення і методика розрахунку ректифікаційних апаратів є подібними до розглянутих вище.
Відзначимо специфічні особливості пристрою розділювальних апаратів для газових сумішей на прикладі ректифікації рідкого повітря, одержуваного методами глибокого охолоджування. Розділення повітря здійснюють в одноколонних розділювальних апаратах, або в апаратах одинарної ректифікації, двоколонних апаратах, або в апаратах подвійної ректифікації.
Установки одинарної ректифікації. Стиснене в компресорі повітря після очищення від пилу, двоокису вуглецю і водяної пари надходить в теплообмінник 1 (рис. 3.31), де охолоджується продуктами ректифікації (киснем і азотом). Потім повітря надходить в змійовик кип’ятильника 2 колони, девін частково конденсується, віддаючи тепло рідкому кисню, що кипить ззовні змійовика. Пара практично чистого кисню відводиться з кип’ятильника в теплообмінник 1.
Частково сконденсоване повітря, що пройшло через дросельний вентиль 3, ще більше охолоджується. Суміш рідкого і пароподібного повітря надходить на верхню тарілку ректифікаційної колони 4. На тарілках колони відбувається звичайний процес ректифікації: під час багатократної взаємодії стікаючої рідини з парою, що піднімаються знизу, і з якої конденсується кисень (висококиплячий компонент), а з рідини випаровується азот (низькокиплячий компонент). У результаті з верхньої частини колони видаляється пара азоту, що приблизно є рівноважною з повітрям, яке подають в колону і яке містить домішки кисню (не більше 7 – 10 %). До кип’ятильника колони надходить чистий кисень. Як вказувалося, кисень і технічний азот прямують в теплообмінник 1 для охолоджування стисненого в компресорі повітря.
Особливістю пристрою ректифікаційної колони 4 є те, що вона не має дефлегматора і працює як вичерпна колона. Це пояснюється тим, що практично неможливо підібрати охолоджувальний агент для конденсації пари дистиляту (азоту), оскільки для цієї мети знадобилася б рідина, що має температуру, нижчу від температури рідкого азоту. Крім того, як початкова суміш і флегма в колону надходить повітря з дуже низькою температурою, за якої точка перетину робочих ліній може практично відповідати складу дистиляту, що взагалі усуває потребу в дефлегматорі.
Істотним недоліком одинарної ректифікації є втрати кисню з азотом. Приблизно третина кисню видаляється з азотом, забруднюючи його, і тільки дві третини кисню, який знаходиться в повітрі і стискається в компресорі, корисно використовується.
Принципово можливим способом підвищення ступеня чистоти азоту і збільшення виходу кисню під час розділення повітря є живлення ректифікаційної колони початковою сумішшю, багатшою на азот, ніж звичайне повітря. Цей принцип використовують в установках подвійної ректифікації для розділення повітря.
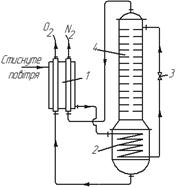
Рис. 3.31. Установка одинарної ректифікації для розділення рідкого повітря:
1 – теплообмінник; 2 – змійовик-кип ’ ятильник; 3 – дросельный вентиль;
4 – ректифікаційна колона
Установки подвійної ректифікації. У такій установці (рис. 3.32) для попереднього збагачення повітря використовують додаткову нижню колону 1, що працює під високим тиском, більшим, ніж тиск в основній верхній колоні 2, яку встановлюють безпосередньо на колоні 1. Завдяки вищому тиску в нижній колоні вона має дефлегматор (охолоджуваний рідким киснем, що стікає з колони 2), який одночасно є кип’ятильником дляколони 2. Вихідне очищене і охолоджене повітря, стиснене до ~7 aт, вводять в змійовик 3 кип’ятильника колони 1. Віддаючитепло, необхідне для кипіння рідини в кип’ятильнику, повітря конденсується. Зріджене повітря проходить через дросельний вентиль 5, охолоджується ще більше і надходить на живлячу тарілку колони 1, в якій підтримується тиск, що дорівнює 6 aт. У результаті в колоні 1 збирається рідина, збагачена ВК (киснем), і в кип’ятильник 4 стікає рідина, що містить приблизно 40–60 % О2. Тут вона частково випаровується унаслідок теплообміну з повітрям, що проходить змійовиком 3. Пара, що утворилася, піднімається вгору і, взаємодіючи із стікаючою рідиною, збагачується азотом. Пара азоту, що містить 94–96 % N2, надходить в трубки дефлегматора, де вона повністю конденсується і віддає тепло рідкому кисню, що стікає колоною 2, і кисню, що кипить в міжтрубному просторі дефлегматора.
Для здійснення процесу теплообміну в дефлегматорі температура кипіння НК (азоту) в трубках дефлегматора повинна бути вищою від температури кипіння кисню в кип’ятильнику колони 2. Це досягається у разі вказаного вище підвищення тиску до ~ 6 am в колоні 1 порівняно з тиском в колоні 2, що дорівнює ~ 1,5 am.
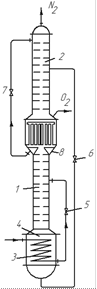
|
| Рис. 3.32.Установка подвійної ректифікації для розділення повітря: 1 – нижня ректифікаційна колона; 2 – верхня ректифікаційна колона; 3 – змійовик; 4 – кип ’ ятильник нижньої колони; 5–7 – дросельні вентилі; 8 – кишеня |
Збагачена киснем рідина з кип’ятильника нижньої колони надходить через дросельний вентиль 6 (знижуючий її тиск до ~1,5 am) на тарілку живлення верхньої колони 2.
Рідкий азот (з концентрацією 98 % N2), сконденсований в дефлегматорі, ділять на дві частини. Майже половину його кількості подають на зрошування колони 1 для повнішого очищення кисню від азоту, а решта його частини, що збирається в кишені 8, через дросельний вентиль надходить як флегма на зрошування колони 2.
Одержувані азот і кисень містять деяку кількість аргону та інших рідкісних газів, які знаходяться у вихідному повітрі. Для підвищення ступеня чистоти кінцевих продуктів розділення доводиться видаляти частину пари з тієї тарілки колони 1, на якій в найбільшій кількості нагро-
маджується аргон. Подальше розділення рідкісних газів здійснюється низькотемпературною ректифікацією в окремих колонних апаратах.
З верхньої частини колони 2 виводять пару азоту, що містить 99,8–99,9 % N2, знизу колони 1 – технічний рідкий кисень (99,3 % О2).
Відомо також інший спосіб підвищення ступеня чистоти азоту у разі використання апарату, в якому замість додаткової колони задіюють дефлегматор з довгими трубками. Охолоджене повітря з компресора частково конденсується і збагачується азотом. Таке попереднє розділення повітря в дефлегматорі, що працює за вищого тиску, ніж колона 2, дає змогу замінити ним колону 1.
Продуктивність існуючих установок для розділення повітря становить до 7500 м3/год повітря і більше.
Питання до розділу ІІІ
1. Загальні відомості про процес перегонки і ректифікації.
2. Характеристика двофазних систем “рідина – рідина”.
3. Ідеальні суміші. Закон Рауля.
4. Проста перегонка. Види перегонки.
5. Ректифікація. Принципи ректифікації. Схема і принцип роботи безперервно діючої ректифікаційної установки.
6. Класифікація бінарних систем ідеальної і реальної суміші. Рівноважний стан.
7. Схема і принцип роботи періодично діючої ректифікаційної колони.
8. Періодична ректифікація бінарних систем, тарілчасті колони.
9. Рівноважна і робочі лінії процесу ректифікації. Побудова робочих ліній.
10. Матеріальний баланс періодично діючої ректифікаційної колони.
11. Тепловий баланс процесу ректифікації.
12. Залежність між флегмовим числом, витратою теплової енергії на процес і розмірами колони.
13. Конструкція ректифікаційних колон. Загальні відомості.
14. Ректифікація багатокомпонентних сумішей.
15. Спеціальні методи перегонки. Азеотропна і екстракційна ректифікації.
16. Молекулярна дистиляція.
17. Порядок розрахунку тарілчастих і насадкових ректифікаційних колон.
18. Фазова рівновага. Правило фаз.
19. Ідеальні суміші.
20. Р – х діаграма ідеальних сумішей.
21. Діаграма t – х – у (залежність температур кипіння і конденсації від складу фаз).
22. Діаграми рівноваги “пара – рідина” (у – х діаграма).
23. Р – х діаграма для сумішей з позитивним відхиленням від закону Рауля.
24. Фазові діаграми азеотропних сумішей.
25. Суміші взаємно нерозчинних рідин.
26. Фракційна перегонка.
27. Проста перегонка з дефлегмацією.
28. Перегонка з водяною парою.
29. Безперервно діюча ректифікаційна установка.
30. Правило Трутона.
31. Розрахунок мінімального флегмового числа.
32. Розрахунок реального флегмового числа.
33. Періодична ректифікація бінарних сумішей.
34. Складові частини ректифікаційної установки.
35. Барботажні колони.
36. Насадкові колони.
37. Плівкові колони.
38. Розрахунок насадкових колон.
39. Розрахунок тарілчастих колон.
40. Розрахунок ректифікації бінарних систем за допомогою ентальпійної діаграми.
41. Низькотемпературна ректифікація.
42. Установки подвійної ректифікації.
|
|
|
|
|
Дата добавления: 2014-11-20; Просмотров: 1707; Нарушение авторских прав?; Мы поможем в написании вашей работы!