КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Лекция 13. Оптические методы определения экотоксикантов
|
|
|
|
Развитие методов определения экотоксикантов, как правило, направлено на увеличение их чувствительности, точности, специфичности и воспроизводимости, а также на упрощение техники измерений. При выборе наиболее подходящего метода руководствуются следующими критериями: 1) способность обеспечивать адекватное измерение аналитического сигнала определяемого соединения; 2) чувствительность, рабочий диапазон концентраций, предел обнаружения, информативность; 3) влияние мешающих компонентов и факторов; 4) возможность автоматизации.
В табл. 13.1 приведены диапазоны рабочих концентраций для наиболее часто применяемых методов. Видно, что большинство из них с успехом можно использовать в диапазоне от 1 мкг/л до 100 мкг/л и выше. Для определения более низких концентраций необходимо предварительное концентрирование определяемых компонентов или их отделение из препарата.
Таблица 13.1. Пределы чувствительности аналитических методов
| Метод | Пределы обнаружения |
| Масс-спектрометрия | 20 пг/мл - 3 мкг/мл |
| Флуориметрия | 5 нг/мл – 3 мкг/мл |
| Хроматография, детектор иони зационного захвата | 30 пг/мл – 1 мг/мл |
| Хроматография, пламенно-ионизационный детектор | 3 нг/мл – 3 мкг/мл |
| Хроматография,инверсионная вольтамперометрия | 3 нг/мл – 3 мкг/мл |
| Иммунохимические методы | 30 пг/мл – 30 нг/мл |
| ИСП-АЭС | 3 нг/мл – 3 мкг/мл |
| ААС с электротермической атомизацией | 7 нг/мл – 3 мкг/мл |
| Активационные методы | 50 пг/мл - 50 нг/мл |
Для контроля экотоксикантов используются все современные высокочувствительные методы аналитической химии. Так, при определении низких содержаний ионов высокотоксичных металлов в основном применяются методы оптической спектроскопии и люминесценции (атомно-эмиссионная спектроскопия с возбуждением от высокочастотного плазменного факела (ИСП-АЭС), атомно-абсорбционная спектроскопия (ААС) с электротермической атомизацией и др.), а также инверсионная вольтамперометрия (ИВА) с химически модифицированными электродами. Для определения органических загрязнителей наряду с хроматогра-фией наблюдается тенденция к более широкому использованию хромато-масс-спектрометрии, иммунохимических и флуоресцентных методов.
|
|
|
Атомная эмиссионная спектроскопия. В основе атомного спектрального анализа лежит индивидуальность спектров испускания и поглощения химических элементов. Атомный эмиссионный спектральный анализ – метод, основанный на изучении спектра паров исследуемого вещества. Наличие в спектре характерных линий, присущих атомам данного элемента, свидетельствует о присутствии этого элемента в анализируемом объекте (качественный анализ). Интенсивность линий спектров элементов служит мерой концентрации последних (количественный анализ).
Процесс возникновения спектральных линий был объяснен Нильсом Бором в его теории строения атома (1913 г.). За исследования в этой области Бор в 1922 г. был удостоен звания лауреата Нобелевской премии. По Бору у атомов водорода электрон, вращающийся на орбите вокруг ядра, обладает постоянным запасом энергии:
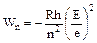
где Wn – энергия на n-ной орбите; Е – заряд ядра; R – постоянная величина, т.н. константа Ридберга (для водорода R = 1,0968×105); h – постоянная Планка; е – заряд электрона; n – квантовое число.
В теории атома Бор показал, что планетарная структура атома и свойства его спектра излучения могут быть объяснены, если считать, что движение электрона подчинено некоторым дополнительным ограничениям – так называемым постулатам Бора. Согласно этим постулатам, для электрона существуют избранные, или «разрешенные» орбиты, двигаясь по которым, он, вопреки законам классической электродинамики, не излучает энергии, но может скачком перейти на более близкую к ядру «дозволенную» орбиту и при этом испустить квант (порцию) электромагнитной энергии, пропорциональный частоте электромагнитной волны.
|
|
|
В современной спектроскопии вместо электронных орбит пользуются часто понятием об уровне энергии атома. На ближайшей к ядру орбите энергия электрона условно считается равной нулю. Чем дальше орбита электрона от ядра, тем больше запас его энергии. Если вещество нагревать, подвергая действию электрических искр (что и применяют в одном из вариантов возбуждения спектров в практическом анализе), происходит переход электронов атома с орбит или уровней, более близких к ядру, на более удаленные. При этом энергия атома увеличивается – атом возбуждается.
Возбужденным называется атом, у которого электроны располагаются на более удаленных орбитах, т.е. обладающий избыточной энергией.
Возбужденные атомы очень неустойчивы, и их электроны легко переходят с более удаленных на более близкие орбиты. Этот переход называется переходом в нормальное состояние и сопровождается выделением энергии в виде лучистой энергии определенной длины волны и определенной частоты колебаний. Если электрон с о-уровня переходит на n-уровень, то при этом:
W0 – Wn = hn
где W0 – энергия электрона на о-уровне; Wn – энергия электрона на n-уровне; h – постоянная Планка; n – частота излучаемых колебаний.
Частота излучаемых колебаний связана с длиной волны уравнением:
l = C/n
где С – скорость света. Вместо частоты n в спектроскопии часто употребляют пропорциональные ей волновые числа
n = 108/l
Возбуждаясь, электрон может переходить на любой из уровней и, в предельном случае, может вообще оторваться от атома. При обратном переходе в нормальное состояние электрон может сразу переходить на нормальный ближайший уровень, или этот переход происходит постепенно – скачками с одного уровня на другой. Когда электрон с более высоких энергетических уровней переходит на более низкий, например, на первый, получаем ряд спектральных линий определенной длины волны и определенной частоты колебаний.
Спектральные линии, возникающие при переходе электрона на один и тот же энергетический уровень, составляют серию спектральных линий.
|
|
|
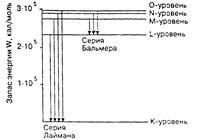
Рис. 13.1. Схема перехода электрона.
Спектр данной серии изображен на рис.13.1 (серия Лаймана). Серия начинается наиболее интенсивными спектральными линиями, соответствующими переходу электрона на данный уровень с наиболее близких уровней. Дальше линии становятся все ближе друг к другу и сливаются в конце концов в широкую полосу с резким окончанием, соответствующим переходу электрона на данный уровень из бесконечности. Рассмотренная серия спектральных линий водорода, называемая серией Лаймана, лежит в далекой ультрафиолетовой области спектра и обнаруживается только на фотопластинках.
Таким образом, возбуждаясь, атомы излучают энергию, которая может быть зафиксирована в виде спектра линий, причем для каждого элемента (например, металла) характерен свой, только ему одному присущий спектр. Благодаря этому можно различить элементы между собой, что является основой качественного спектрального анализа. Классификация методов атомной спектроскопии представлена в табл. 13.1.
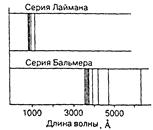
Рис. 13.2. Серии спектральных линий водорода.
Таблица 13.1. Классификация основных методов атомной спектроскопии
| Метод | Диапазон электромагнитного излучения | Процесс | Способ | ||
| атомизации | возбуждения | регистрации | |||
| Атомно-эмис-сионный (АЭС) | Оптический | Эмиссия (фотонов) | Высокотемпературный | Высокотемпературный | Электромагнитная |
| Атомно-флуо-ресцентный (АФС) | « | « | « | ЭМИ (УФ-вид.) | « |
| Атомно-абсорб-ционный (ААС) | « | Абсорбция (фотонов) | « | Не требуется | « |
| Рентгеноэмиссионный (РЭА) | Рентгеновский | Эмиссия (фотонов) | Не требуется | Ноток электронов | « |
| Рентгенофлуоресцентный (РФА) | « | « | « | ЭМИ (рентг.) | « |
| Рентгеноабсорбционный (РАА) | « | Абсорбция (фотонов) | « | Не требуется | « |
| Рентгеновский фотоэлектронный (РФЭС) | Регистрация электронного спектра | Эмиссия (электронов) | « | ЭМИ (рентг.) | Электронная |
| Оже-элект-ронный (ОЭС) | с кинетич. энергией электронов до 1500 эВ | « | « | Поток электронов | « |
Источники возбуждения спектров. Для получения спектра следует «возбудить» атомы. В практике атомно = эмиссионного спектрального анализа (АЭС) в качестве источников возбуждения спектров применяют пламя, электрические дуги постоянного и переменного тока, низко- и высоковольтную конденсированную искру, низковольтный импульсный разряд, различные формы тлеющего газового разряда и др. В последние годы начинают широко использовать также различные виды высокочастотных разрядов — источник индуктивно-связанной высокочастотной плазмы (ИСП), микроволновой разряд, лазеры и др. Основные типы атомизаторов в АЭС представлены в табл. 13.2.
|
|
|
Пламя используют в качестве источника света в методе фотометрии пламени, а также как один из основных способов атомизации веществ в методе атомно-абсорбционного анализа. В зависимости от состава горючей смеси (воздух-пропан, воздух-ацетилен, воздух-водород и др.) температура пламени может поддерживаться в интервале 2000-3000 К, что обеспечивает достаточно низкий Сн обнаружения элементов (0,001-1 мг/л).
Электрическая дута постоянного тока – более высокотемпературный источник, чем пламя. Анализируемый образец в измельченном виде помещают в углубление в нижнем электроде. Температура плазмы дуги зависит от материала электродов, наиболее высокой она будет в случае применения угольных электродов (около 7000 К). Подобные источники света применялись ранее в кинопроекторах в больших кинотеатрах.
Таблица 13.2. Основные типы атомизаторов в АЭС
| Тип источника атомизаиии | Т,°С | Состояние пробы | Сmin, % масс. | Sr |
| Пламя | 1500-3000 | Раствор | 10-7-10-2 | 0,01-0.05 |
| Электрическая дуга | 3000-7000 | Твердая | 10-4-10-2 | 0,1-0,2 |
| Электрическая искра | 10000-12000 | Твердая | 10-3-10-1 | 0,05-0,10 |
| Индуктивно связанная плазма | 6000-10000 | Раствор | 10-8-10-2 | 0,01-0,05 |
В дуге постоянного тока можно одновременно определять десятки элементов, но точность метода невысока из-за нестабильности разряда.
Более стабильные условия возбуждения создает дуга переменного тока. В современных генераторах дуги переменного тока можно получить различные режимы возбуждения: низковольтную искру, высокочастотную искру, дугу переменного тока, импульсный разряд и др. Такие источники света с различными режимами используют при определении металлов и трудновозбудимых элементов (углерод, галогены, газы, содержащиеся в металлах и др.).
Высоковольтная конденсированная искра служит главным образом источником света при анализе металлов. Стабильность искрового разряда позволяет получить высокую воспроизводимость анализа, однако сложные процессы, происходящие на поверхности анализируемых электродов, приводит к изменениям состава плазмы разряда.
В последние годы разработаны новые источники возбуждения спектров – с помощьювысокочастотных плазмотронов, лазера и некоторых формтлеющего разряда.
Для получения индуктивно-связанной плазмы (ИСП) используют ВЧ-генераторы с рабочей частотой в диапазоне 27-50 МГц. Плазма образуется в результате индукционного нагрева газа (чаще всего аргона), протекающего через систему концентрических трубок, размещенных внутри рабочей катушки ВЧ-генератора (рис. 13.3). Плазмотрон изготавливается из кварца, а во избежание разрушения горелки при высокой температуре ее охлаждают дополнительным внешним потоком газа.
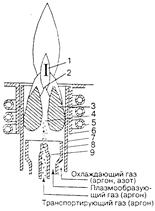
| Рис. 13.3. Схема горелки для высокочастотного индукционного разряда. Газовые потоки и зоны в факеле плазмы: 1 – аналитическая зона; 2 – зона первичного излучения; 3 – зона разряда (скин-слой); 4 – центральный канал (зона предварительного нагрева); 5 – индуктор; 6 – защитная трубка, предотвращающая пробой на индуктор; 7 – внешняя трубка; 8 – промежуточная трубка; 9 – центральная трубка. |
В поперечном сечении разряд имеет форму тора. Пробу в виде аэрозоля подают по центральной трубке горелки в осевую зону разряда. Аэрозоль проходит по центральному каналу разряда, не задевая электропроводящего скин-слоя и не влияя на его характеристики. В этом заключается одна из главных особенностей ИСП-разряда, отличающая его, например, от дуговых плазмотронов.
Электронная температура разряда 6000-10000 К, т.е. существенно выше, чем в дуге или пламени. Продолжительность пребывания частичек аэрозоля в наиболее горячей зоне составляет примерно 0,01 с, что обеспечивает их полное испарение, эффективную атомизацию и возбуждение. Плазма стабильна в пространстве и времени.
Возбуждение спектров в ИСП-разряде позволяет определять содержания примерно 70-ти элементов Периодической системы, включая и такие, как фосфор, сера, бор, мышьяк и олово. Интервал определяемых концентраций 10-1-10-4 мг/л при относительном стандартном отклонении ~0,01, причем градуировочные графики линейны в пределах 4-6 порядков концентрации. ИСП – самый современный источник атомизации, обладающий по целому ряду показателей наилучшими аналитическими возможностями и метрологическими характеристиками.
Атомы анализируемого вещества могут поступать в разряд не только в результате термического испарения, но и под действием бомбардировки поверхности анализируемого вещества ионами. В спектральном анализе для этой цели используют тлеющий разряд постоянного тока при пониженном давлении инертного газа, осуществляемый в специальных разрядных трубках с «полым катодом». Этим способом можно успешно определять такие элементы, как углерод, серу и фосфор, а также многие металлы.
Любой метод оптической спектроскопии может быть существенно улучшен при использовании лазеров. Прямое испарение пробы, например, СО2-лазером, позволяет в одном эксперименте определять несколько десятков элементов, а высокая монохроматичность лазерного излучения дает возможность избирательно анализировать ультрамалые количества вещества в атмосфере.
Спектральные приборы. Основными частями спектрального прибора (рис. 13.4, а) являются: входная щель S, освещаемая исследуемым излучением; объектив коллиматора О1, в фокальной плоскости которого расположена щель S; диспергирующее устройство D, работающее в параллельных пучках лучей: фокусирующий объектив О2, создающий в своей фокальной поверхности Р монохроматические изображения входной щели, совокупность которых и образует спектр. В качестве диспергирующего элемента используют, как правило, либо призмы, либо дифракционные решетки. Схема эмиссионного спектрометра представлена на рис. 13.4, б.
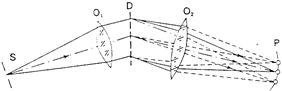
| Рис. 13.4, а. Принципиальная оптическая схема спектрального прибора. |
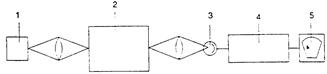
Рис. 13.4, б. Схема однолучевого эмиссионного спектрометра: l – проба; 2 – устройство выделения спектрального интервала (монохроматор, фильтр); 3 – фотоприемник; 4 – усилитель; 5 – индикатор выходного сигнала.
Действие призмы как диспергирующего элемента спектрального прибора основано на зависимости показателя преломления материала призмы от длины излучения. Разрешающая способность призмы определяется ее размером и материалом. Исследование все более сложных спектров потребовало увеличения разрешающей силы приборов, т.е. повышения способности к различению соседних спектральных линий. Сначала этого повышения добивались увеличением числа призм. Но призмы сильно поглощают свет в той области, где дисперсия высока, и прозрачны там, где дисперсия мала.
Поэтому возникала необходимость в новых диспергирующих элементах. Фраунгофер предложил для этой цели дифракционную решетку. Спектр, даваемый дифракционной решеткой, возникает вследствие дифракции света, проходящего через систему очень тонких щелей, и последующей интерференции дифрагированных лучей в точке наблюдения. Последовательность линий в спектре, даваемом дифракционной решеткой, противоположна последовательности линий в спектре призмы. В современных спектральных приборах с высокой разрешающей способностью применяются главным образом отражательные дифракционные решетки с максимальным количеством штрихов (щелей) на 1 мм в интервале 2000-2400.
Отечественная промышленность выпускает целый ряд призменных и дифракционных спектральных приборов. К простейшим из них относятся стилоскопы и стилометры для визуального спектрального анализа, которые используют для экспресс-анализа сталей и сплавов в заводских лабораториях металлургических заводов.
Для более детальных анализов с использованием фотографических методов регистрации спектров широкое распространение получили призменные спектрографы с кварцевой оптикой ИСП-28 и ИСП-30 (рабочая область спектра 200-600 нм). Они позволяют различать спектральные линии, отстоящие друг от друга на расстоянии не менее 0,03 нм. Оптическая схема одного из наиболее популярных спектрографов для УФ-области спектра приведена на рис. 13.5.
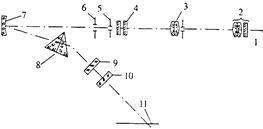
| Рис. 13.5. Схема спектрографа ИСП-28: 1 – источник света; 2, 3 и 4 – трехлинзовый конденсор; 5 – щель; 6 – диафрагма; 7 – зеркальный объектив коллиматора; 8 – диспергирующая призма; 9 и 10 – линзы объектива камеры; 11 – плоскость спектра. |
Из дифракционных спектрографов чаще других применяют приборы с плоской решеткой и зеркальной фокусирующей оптикой. По такой схеме построены спектрографы ДФС-8 и ДФС-13. Они имеют сменные дифракционные решетки с 600 и 1200 штрих/мм и рабочую область спектра от 200 до 1000 нм.
Наиболее широко распространены фотоэлектрические спектрометры (полихроматоры, или квантометры) с фотоэлектрическим способом регистрации спектров. Эти приборы имеют на выходе щель, на которую последовательно выводят аналитические линии всех определяемых элементов, что ограничивает скорость анализа. Для одновременного определения содержания всех элементов в анализируемой пробе необходимо выделить из спектра соответствующее число линий разных элементов. Для этого в фокальной плоскости квантометра устанавливают соответствующее число выходных щелей.
В первом отечественном квантометреДФС-10 полихроматор снабжен 36-ю передвижными щелями, позволяющими одновременно выделять из спектра 36 спектральных линий. Прибор имеет рабочую область спектра 170–700 нм. В качестве диспергирующего элемента использована дифракционная решетка с 1200 или 1800 штрих/мм и радиусом кривизны 2 м. Входная щель, дифракционная решетка и выходные щели размещены по кругу Роуланда (диаметр круга равен радиусу кривизны решетки). Прибор позволяет выполнять анализы по 12 различным программам, причем число определяемых элементов по каждой из программ можно варьировать от 1 до 35. Для одновременного определения 10 элементов в одном образце требуется не более 2 мин.
В последние два десятилетия интенсивно развивается метод АЭС со сравнительно новьм источником возбуждения – высокочастотным плазмотроном (ИСП). Принцип действия плазмотрона состоит в том, что при пропускании через индукционную катушку, соединенную с высокочастотным генератором, инертного газа (аргона, гелия) последний ионизируется, и на выходе горелки образуется плазменный факел с температурой, достигающей 10 000 К. Анализируемый образец вводят в пламя горелки с потоком инертного газа, причем присутствие легкоионизирующихся элементов практически не влияет на режим ее работы. Опыт эксплуатации приборов с высокочастотным индукционным разрядом подтверждает, что аналитические характеристики этого источника возбуждения спектров эмиссии выше других. С помощью ИСП-АЭС – приборов получаются более точные результаты, так как практически отсутствует влияние матричных элементов, а интенсивность фона в 100 и более раз меньше. Особенно важно, что градуировочные графики линейны в диапазоне четырех порядков. Пределы обнаружения большинства элементов по сравнению с другими источниками возбуждения ниже на 1-3 порядка (табл. 13.2) Следует заметить, что наиболее эффективной областью применения метода ИСП-АЭС является анализ воды
При анализе высокоминерализованных вод следует учитывать рассеяние света, вызываемое кальцием и магнием. В отличие от других источников возбуждения в случае ИСП наличие хлорида натрия практически не влияет на пределы обнаружения большинства элементов. Однако влияние препарата в АЭС больше, чем в атомной абсорбции.
Таблица 13.2. Пределы обнаружения некоторых высокотоксичных элементов методом ИСП-АЭС и верхние границы линейности градуировочных графиков для проб воды, мг/л
| Элемент | Предел обнаружения | Верхняя граница линейности | Элемент | Предел обнаружения | Верхняя граница линейности |
| As | 0,050 | Pb | 0,025 | ||
| Be | 0,001 | Se | 0,050 | ||
| Cd | 0,002 | Th | 0,030 | ||
| Cr | 0,005 | Tl | 0,060 | ||
| Cu | 0,002 | U | 0,100 | ||
| Ni | 0,010 | Zn | 0,004 |
Атомная абсорбционная спектроскопия. Атомно-абсорбционная спектроскопия (ААС, атомно-абсорбционный анализ) – метод элементного анализа вещества по атомным спектрам поглощения. Для наблюдения этих спектров через атомный пар пробы пропускают видимое или УФ-излучение. В результате поглощения квантов излучения электроны атомов переходят с нижних энергетических уровней на возбужденные. Этим переходам в атомном спектре соответствуют так называемые резонансные линии, характерные для данного элемента.
В атомно-абсорбционной спектрометрии измеряют светопоглощение в газовой фазе при высоких температурах (в пламени), обусловленное незаряженными, невозбужденными свободными атомами. Отношение количества атомов какого-либо элемента в возбужденном (Na) и основном (N0) состояниях описывается распределением Больцмана:
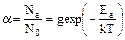
где g – статистический множитель; Еa – энергия возбуждения; k – константа Больцмана (k = R / NA); Т – абсолютная температура.
При обычных температурах пламени (2000-3000°С) величина а очень мала. Поэтому величину N0 можно считать постоянной, равной концентрации атомов. Следовательно, оптическая плотность атомного пара, измеренная при его резонансной частоте поглощения, пропорциональна общей концентрации определяемого вещества.
При атомно-абсорбционных измерениях частота падающего света должна строго соответствовать резонансной частоте поглощения атомов. Поэтому источники непрерывного спектра здесь неприменимы. В качестве источников в атомной абсорбции применяют специальные лампы с полым катодом, изготовленным из определяемого металла. Напряжение питания таких ламп достигает 400 В, сила тока до 100 мА. Лампы с полым катодом достаточно дороги, однако при их использовании достигается абсолютная селективность. Сигнал, обусловленный собственным излучением возбужденных атомов в пламени, можно исключить, применяя модуляцию источника излучения (налагая на катод лампы переменное напряжение). В противном случае измеренные значения оптических плотностей окажутся заниженными (рис. 13.6).
Измерения в атомно-абсорбционном методе основаны на законе Ламберта–Бера. Здесь также необходимо предварительно построение градуировочной кривой для каждого определяемого элемента. Погрешность определения составляет около 2%, чувствительность (предел обнаружения) не менее 1 мкг/мл, в отдельных случаях до 0,005 мкг/мл. Метод ААС – один из лучших способов определения металлов в экологических пробах. Атомно-абсорбционным методом можно определять и некоторые неметаллы (В, Si, As, Se, Те).
Исследуемое вещество атомизируют, распыляя его раствор в пламя газовой горелки (разновидность фотометрии пламени) или испаряя сухой остаток раствора в электрической трубчатой печи при температурах до 3000°С. Обычно через атомный пар пропускают линейчатое излучение, соответствующее атомному спектру определяемого элемента.
В качестве источников излучения используют лампы с полым катодом или безэлектродные радиочастотные лампы. Световой поток после прохождения через поглощающий слой и монохроматор, выделяющий резонансную линию, регистрируют фотоэлектрически.
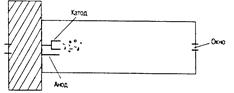
Рис. 13.6. Лампа с полым катодом
В соответствии с законом Бугера–Ламберта–Бера мерой концентрации определяемого элемента служит поглощающая способность вещества. Достоинства метода атомно-абсорбционной спектроскопии: высокая избирательность определения индивидуальных элементов, низкие значения Сн (10–1 и 10–4 мг/л в случае использования газовой горелки и графитовой печи соответственно), хорошая воспроизводимость (относительное стандартное отклонение ~0,01) и большая производительность (до 500 определений в 1 ч).
Атомизация в пламенах. Принципиальная схема атомно-абсорбционного спектрофотометра показана на рис..д. Свет от источника резонансного излучения 1 пропускают через пламя, в которое впрыскивается мелкодисперсный аэрозоль 10 раствора пробы. Излучение резонансной линии выделяют из спектра с помощью монохроматора 2 и направляют на фотоэлектрический детектор 3 (обычно фотоумножитель). Выходной сигнал детектора после усиления (4) регистрируют гальванометром 5, цифровым вольтметром или записывают в аналоговой форме на ленте пишущего потенциометра (6).
Интенсивность резонансного излучения измеряют дважды – до распыления анализируемого образца в пламя и в момент его распыления. Разность этих двух отсчетов и определяет значение аналитического сигнала.
Для получения пламени используют различные комбинации горючих газов с окислителями, например, водорода, пропана или ацетилена с воздухом или оксидом азота. В практике атомно-абсорбционного анализа чаще всего применяют воздушно-ацетиленовое пламя.
Рис. 13.7. Блок-с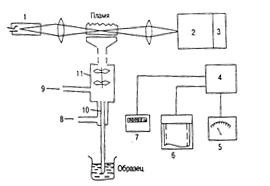
хема атомно-абсорбционного спектрометра: 1 – линейчатый источник резонансного излучения; 2 – монохроматор; 3 – детектор; 4 – усилитель; 5 – стрелочный прибор; 6 – самописец; 7 – цифропечатающее устройство; 8, 9 – ввод окислителя и топлива соответственно; 10 – распылитель; 11 – распылительная камера.
Его используют для определения щелочных и шелочно-земельных элементов, а также таких металлов, как хром, железо, кобальт, никель, магний, молибден, стронций, благородные металлы и др. В воздушно-ацетиленовом пламени нельзя определять (слишком высокая энергия связи металл–кислород) алюминий, тантал, титан, цирконий и др.
Пламя ацетилена с воздухом обладает высокой прозрачностью в области длин волн более 200 нм, слабой собственной эмиссией и обеспечивает высокую эффективность атомизации более чем 30-ти элементов. Пламя ацетилена и оксида азота (I) имеет почти на 900 К более высокую температуру и в нем создаются условия для атомизации значительно более широкого круга элементов и их соединений. Пламя отличается высокой прозрачностью во всем интервале длин волн, используемых в атомно-абсорбционном анализе (190-850 нм).
Эти две газовые смеси взаимно дополняют друг друга и совместно позволяют определять примерно 70 элементов, многие из которых являются приоритетными загрязнителями окружающей среды (хром, кобальт, никель, молибден, стронций, олово, свинец, кадмий и др.).
Электротермическая атомизация. Атомизация в пламени имеет ряд серьезных ограничений, обусловленных химическими реакциями в пламени и малой продолжительностью пребывания в нем частиц определяемого вещества (~10–3 с). Более дешевыми, безопасными и эффективными оказались электротермические атомизаторы, некоторые из которых успешно применяются в практическом атомно-абсорбционном анализе.
При использовании атомизатора типа графитовой кюветы анализируемую пробу в виде раствора наносят на торец угольного электрода и после высушивания капельки электрод вводят через отверстие в предварительно разогретую до 2300 К кювету (графитовая трубка длиной около 5 см и внутренним диаметром 4-5 мм). При соприкосновении электрода с кюветой происходит дополнительный электроконтактный разогрев электрода, и проба в течение нескольких долей секунды испаряется внутрь кюветы.
Для атомизации пробы используют и тонкостенную графитовую печь. Анализируемую пробу в виде раствора дозируют.микропипеткой (5-100 мкл) через отверстие на стенку холодной печи. Печь постоянно обдувается потоком аргона, что предохраняет ее от обгорания и способствует удалению испаренной пробы из атомизатора. После высушивания пробы печь разогревается до температуры 3000 К. При этом сухой остаток пробы испаряется, и пар вещества заполняет всю трубку печи.
Метод атомной абсорбции с применением электротермического атомизатора обеспечивает рекордно низкие значения Сн по многим элементам. Их численные значения колеблются для разных элементов от десятых до десятитысячных долей нанограмма в одном миллилитре раствора, достигая в абсолютном выражении величины 10–12-10–14 г. Столь высокая абсолютная чувствительность метода достигается благодаря импульсному характеру испарения всей пробы и формированию поглощающего слоя атомов в пространстве, ограниченном стенками печи.
Техника анализа. Для измерения сигнала абсорбции необходим внешний источник излучения (возбуждения спектра). Лучшим для этой цели является источник линейчатого спектра: разрядные трубки или лампы с полым катодом и безэлектродные лампы с высокочастотным возбуждением.
Лампа с полым катодом представляет собой герметичный стеклянный баллон с впаянными в него катодом и анодом, а также окном для выхода излучения. Баллон заполнен инертным газом (аргоном или неоном) до давления в несколько гектопаскалей. Катод, в форме цилиндра или стакана, изготовлен из чистого металла или сплава, содержащего требуемый элемент. При подаче на электроды напряжения ~300 В в лампе возникает слаботочный тлеющий разряд. Ионы аргона или неона, бомбардируя поверхность катода, распыляют его, и атомы возбуждаются в газовом разряде посредством столкновений с электронами и ионами. В результате лампа излучает эмиссионный спектр нужного (определяемого) элемента.
Для получения спектров летучих элементов (мышьяк, сурьма, висмут, селен, теллур и др.) лучше подходят безэлектродные лампы с высокочастотным возбуждением (27 МГц и выше). Они представляют собой небольшие кварцевые ампулы, заполненные инертным газом и содержащие примерно 10 мг летучего соединения определяемого элемента. Газовый разряд в безэлектродных лампах происходит в очень тонком слое непосредственно у стенок ампулы (скин-эффект высокочастотного поля).
Меняя лампы (источник излучения), предназначенные для определения конкретных элементов, можно определять до 70 элементов (преимущественно металлов) в различных объектах (воде, почве, продуктах жизнедеятельности организма, нефтях, минералах и др.) на уровне следовых концентраций (до 10–6%).
Наряду с однолучевыми приборами, для измерения атомной абсорбции применяют и двухлучевые спектрофотометры. В России выпускаются атомно-абсорбционные спектрофотометры типов С-115, С-115 MI и ААС-А со спектральным диапазоном 190-900 нм. В качестве атомизаторов применяется пламя и графитовая печь. Приборы комплектуются набором спектральных ламп на 30, 30 и 67 элементов соответственно. Предел обнаружения 0,001 мг/л.
Метод атомно-абсорбционной спектроскопии, в основе которого лежит противоположное применяемому в АЭС явление – поглощение резонансной линии свободными атомами определяемого элемента, находящимися в невозбужденном состоянии, при прохождении света через пары исследуемого образца, состоит в измерении этого поглощения и обладает быстротой и хорошей точностью. Его основное преимущество перед другими методами в высокой селективности, простоте подготовки проб к анализу и возможности определения нескольких элементов из одного раствора по единой методике. Однако при всех достоинствах он уступает по производительности атомно-эмиссионной спектроскопии. Кроме того, как и в методе АЭС, для получения надежных результатов также следует учитывать влияние базовых элементов объекта, что вызывает необходимость использования для градуировки приборов стандартных образцов.
В качестве источника света наиболее распространены лампы с полым катодом, которые представляют собой герметичный баллон из стекла с кварцевым окном, пропускающим ультрафиолетовое излучение. В баллон впаяны два электрода: катод в виде полого цилиндра, изготовленный из металла, для определения которого предназначена лампа, и анод произвольной формы. При подаче на лампу тока силой 5-30 мА при выходном напряжении 300-800 В пары металла, из которого изготовлен катод, поступают в плазму разряда и испускают свет. Реже используются лампы с СВЧ – возбуждением. Спектр таких источников линейчатый, интервал длин волн испускаемого света узкий (порядка 0,001 нм), а линии поглощения определяемых элементов заметно шире, аналитический сигнал можно измерять практически селективно. При этом другие элементы не мешают проведению анализа. Однако для каждого элемента приходится иметь свой источник света.
Для превращения растворов анализируемых веществ в атомный пар чаще всего применяют щелевые горелки длиной 5-10 см. Они довольно однотипны по конструкции и легко заменяются. Большинство приборов рассчитаны на использование в качестве окислителей воздуха, кислорода и закиси азота, а в качестве топлива - пропана, ацетилена и водорода. Наибольшее распространение получило воздушно-ацетиленовое пламя (2200-2400°С), которое позволяет определять многие высокотоксичные металлы (Рb, Cd, Zn, Сu, Cr и др.). Для определения элементов с более высокой температурой парообразования (Al, Be, Mo и др.) широкое признание получила смесь закись азота-ацетилен (3100-3200°С), поскольку она более безопасна в работе, чем смеси с кислородом. Для обнаружения мышьяка и селена в виде гидридов требуется восстановительное пламя, образующееся при сжигании водорода в смеси аргон-воздух.
Наряду с пламенными атомизаторами в ААС в последнее время широко применяются электротермические атомизаторы, имеющие ряд неоспоримых преимуществ, таких как более низкие пределы обнаружения (до 10–12 %), малый объем пробы (1-10 мкл), отсутствие взрывоопасных газов. Метод основан на испарении (атомизации) элементов в графитовой кювете, нагреваемой электрическим током, которая представляет собой графитовую трубку длиной 20-50 мм, внутренним диаметром 3-5 мм и внешним – 5-8 мм. Пробу вводят в кювету через отверстие (2 мм) с помощью микропипетки или автосамплера. Время определения одного элемента составляет 1-2 мин. В этих условиях возможно определение до 0,02 мкг/л кадмия, 1,0 мкг/л свинца, 0,016 мкг/л цинка (табл. 13.3).
Таблица 13.3. Абсолютные и концентрационные пределы обнаружения элементов методом ААС с электротермической атомизацией
| Элемент | Абсолютное количество, мкг | Концентрация, мкг/л | Элемент | Абсолютное количество, мкг | Концентрация, мкг/л |
| As | 1×10–4 | 20,0 | Hg | 1×10–4 | 20,0 |
| Be | 9×10–7 | 0,18 | Ni | 1×10–5 | 2,0 |
| Cd | 1×10–7 | 0,02 | Pb | 5×10–6 | 1,0 |
| Сr | 5×10–6 | 1,0 | Se | 1×10–4 | |
| Сu | 7×10–6 | 1,4 | Zn | 8×10–8 | 0,016 |
Сейчас появились импульсные атомизаторы, которые позволяют получить мгновенные «облака» поглощающих паров высокой концентрации, что обеспечивает повышение чувствительности определений на полпорядка и более в зависимости от природы элемента.
В отличие от рассмотренных выше элементов определение общего содержания ртути методом ААС основано на измерении поглощения света ее парами, при длине волны 253,7 нм в газовой кювете при комнатной температуре («метод холодного пара»). В качестве восстановителей применяют хлорид олова, станнит натрия, аскорбиновую кислоту и др. Предел обнаружения составляет 0,2 мкг/л, диапазон измеряемых концентраций 0,2-10 мкг/л. Кроме того, для определения ртути после ее восстановления хлоридом олова предложен атомно-флуоресцентный метод с применением низкотемпературного пропан-воздушного пламени. Флуоресценция паров ртути возбуждается также излучением ртутной лампы при 184,9 и 253,7 нм. В этом случае предел обнаружения метода достигает 10-8%.
Надежным, экспрессным и высокочувствительным методом контроля экотоксикантов, позволяющим определять как суммарное содержание загрязняющих веществ, так и индивидуальных соединений, является люминесцентный метод анализа. В некоторых случаях предел обнаружения люминесцентных методов сравним с пределом обнаружения радиоактивационного анализа. Наибольшее применение находят фотолюминесцентные методы (флуоресценция и фосфоресценция). В результате химических реакций между реагентом и определяемым веществом образуются флуоресцирующие соединения, по интенсивности свечения которых определяют концентрацию исследуемого компонента. Регистрацию спектров в настоящее время осуществляют исключительно фотоэлектрическим способом.
Это относится и к программе фонового мониторинга природных объектов. Для целей мониторинга ПАУ создан банк спектров при 77 К, который опубликован в виде атласа (при 77 К спектры флуоресценции имеют более узкие и потому более специфичные полосы).. На основе проведенных исследований разработаны высокочувствительные и селективные методы определения ПАУ и их производных в многокомпонентных природных и техногенных системах: в воздухе, почве, растениях, атмосферных осадках, природных и сточных водах, донных отложениях, горных породах, минералах, нефтях, высокотемпературных пиролизатах, отработанных газах автомобильных двигателей, саже и т.д. Предел обнаружения в однокомпонентных растворах для разных соединений находится в диапазоне от 0,01 до 1 нг/мл.
Расчет содержания ПАУ в исследуемых растворах основан на простом соотношении: С1/С2 = I1/I2, где C1 и С2 – концентрации одного и того же вещества; I1 и I2 – соответствующие им интенсивности.
Если в соотношении C2 принять за искомую концентрацию ПАУ (Сх), а C1 – за концентрацию его эталонного раствора (Сэт), то получим:
С х = С х I х/ I эт = КС ст I х/ I ст
Коэффициент К можно оценить по отношению интенсивности линии при 419,2 нм в спектре флуоресценции раствора 1,12-бензперилена в н -гексане (I ст), принятого за единый стандарт, к интенсивности характеристической линии в спектре флуоресценции эталонного раствора определяемого ПАУ (I эт): К = I ст/ I эт.
В отличие от рассмотренных выше методы спектрометрии в ультрафиолет двои, видимой и инфракрасной областях спектра практически не используются в аналитической химии экотоксикантов из-за низкой чувствительности и недостаточной селективности при анализе сложных препаратов. Перспективы этих методов в анализе экотоксикантов связаны с их применением для детектирования в хроматографии. В частности, система газожидкостной хроматограф – инфракрасный фурье-спектрометр широко применяется в повседневных анализах многих высокотоксичных веществ. Сравнение возможностей комбинированных методов анализа при определении хлорнитробензолов в сточных водах показало, что только инфракрасная фурье-спектроскопия в сочетании с хроматографией позволяет различать изомеры данных соединений, хотя она и уступает по чувствительности хромато-масс-спектрометрии на 2-3 порядка.
Рассматривая спектроскопические методы определения и обнаружения экотоксикантов в целом, можно видеть, что между ними существуют принципиальные различия. В абсорбционной спектроскопии измеряется энергия, не поглощенная образцом, а в эмиссионной спектроскопии – энергия, выделяемая в процессах возбуждения исследуемых компонентов, т.е. эмиссионный анализ чувствительнее, ибо здесь интенсивность сигнала пропорциональна количеству вещества.
Следует заметить, что благодаря использованию индуктивно-связан-ной плазмы в качестве источника ионов приих масс-спектрометрическом определении (ИСП-МС) удалось достигнуть исключительно низких пределов обнаружения Применение масс-анализатора позволяет проводить многоэлементный анализ из одной пробы. Пределы обнаружения для большинства металлов находятся в диапазоне от 2 до 30 нг/л.
Инфракрасная спектроскопия. Инфракрасная спектроскопия (ИК-спектроскопия) – раздел спектроскопии, изучающий спектры поглощения (и отражения) электромагнитных волн в ПК-области. Это длинноволновая область спектра, границы которой условны. Она начинается сразу же за красным концом видимого спектра и далеко вклинивается в микроволновую область, граница которой находится около l = 2,5 мм. Для характеристики ИК-спектра вместо длины волны обычно используют волновые числа n в см–1. В этом случае границы ИК-области спектра простираются от 50 до 5000 см–1.
ИК-спектры возникают в результате колебательного (отчасти вращательного) движения молекул, а именно – в результате переходов между колебательными уровнями основного электронного состояния молекул. В отличие от УФ-спектров, при образовании которых молекулы переходят в возбужденное, часто более активное состояние, ИК-спектры получают при более «мягком» возбуждении. В результате для них характерна тонкая структура с гораздо большим числом узких полос, которые более определенно характеризуют молекулу, чем УФ-спектры.
Инфракрасная область спектра была открыта около 1800 г. английским астрономом Уильямом Гершелем, который обнаружил, что термометр, помещенный за красным краем солнечного спектра, показывает заметное повышение температуры. Однако понадобилось свыше ста лет, прежде чем американский физик Кобленц опубликовал в 1905 г. обширный обзор ИК-спектров многих классов органических и неорганических соединений и рассмотрел соответствие между спектрами и структурой. Широкое признание больших возможностей применения ИК-спектроскопии для решения структурных и аналитических задач в органической химии пришло только в начале 40-х гг. В это время были созданы автоматические регистрирующие приборы, которые применяли в работе над некоторыми важными проблемами военного времени, такими, как анализ авиационных топлив, синтетических резин и волокон, выяснение структуры пенициллина.
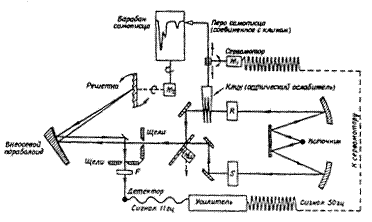
Вскоре появились относительно недорогие, но достаточно хорошие коммерческие приборы, производство которых сильно выросло после 1950 г., и в настоящее время вряд ли найдутся лаборатории, работающие с органическими веществами и не имеющие ИК-спектрометров. Схема спектрофотометра высокого разрешения с дифракционной решеткой представлена на рис..д.
Свет от источника разделяется поровну зеркалами, расположенными определенным образом, на два луча, один из которых проходит через кювету с образцом S, а другой через пустую кювету, или кювету сравнения, R. Затем лучи сводятся с помощью вращающегося полукруглого зеркала, приводимого в движение моторчиком М3, и полученный таким образом единый луч представляет собой чередование (с частотой 11Гц) луча, проходящего через образец, и луча сравнения.
Пульсирующий луч проходит через входную щель монохроматора и затем диспергируется решеткой. Оптический фильтр F пропускает только узкую полосу частот, поступающих из выходной щели и попадающих после прохождения фильтра F на детектор (термопара). Если свет с данной частотой преимущественно поглощается образцом в кювете S, то в детекторе генерируется переменный сигнал (11 Гц). Усиленный в значительной степени этот сигнал идет к сервомотору M1, который перемещает клин луча сравнения до тех пор, пока разность сигналов не уменьшится до нуля. Передвижение клина сопровождается одновременным и пропорциональным перемещением пера самописца, так что по мере развертывания спектра вращением решетки и барабана самописца перо автоматически вычерчивает кривую процента пропускания в зависимости от волнового числа.
 Спектральный диапазон ИК-спектрометров составляет обычно 200–4000 см–1 (разрешение полос до 0,001 см–1). Качественный анализ на основе ИК-спектров возможен (в отличие от УФ-спектров) благодаря их высокой индивидуальности и существованию характеристических колебаний некоторых атомных групп, например, СН3, CN, NО2 и др. (табл...). Это наиболее важные колебания в ИК-спектрах (3700—1500 см–1), называемые областью функциональных групп. Колебательные спектры обладают высокой специфичностью и широко используются для идентификации веществ. Каждому веществу присущ свойственный только ему набор полос и не существует двух веществ, которые имели бы одинаковые колебательные спектры.
Спектральный диапазон ИК-спектрометров составляет обычно 200–4000 см–1 (разрешение полос до 0,001 см–1). Качественный анализ на основе ИК-спектров возможен (в отличие от УФ-спектров) благодаря их высокой индивидуальности и существованию характеристических колебаний некоторых атомных групп, например, СН3, CN, NО2 и др. (табл...). Это наиболее важные колебания в ИК-спектрах (3700—1500 см–1), называемые областью функциональных групп. Колебательные спектры обладают высокой специфичностью и широко используются для идентификации веществ. Каждому веществу присущ свойственный только ему набор полос и не существует двух веществ, которые имели бы одинаковые колебательные спектры.
Рис. 13.8. Схема инфракрасного двухлучевого спектрофотометра с дифракционной решеткой. M1 – мотор, перемещающий оптический клин и перо самописца; М2 – мотор, вращающий решетку и барабан самописца; М3 – мотор, вращающий зеркальный прерыватель света; S – кювета с образцом; R – сравнительная кювета; F – оптический фильтр, поглощающий полностью всю ту область частот, которая является результатом дифракции более высоких порядков и которая при отсутствии фильтра накладывалась бы на измеряемую частоту.
В настоящее время имеются атласы ИК-спектров для различных классов органических, элементорганических и неорганических веществ, в которых указаны условия подготовки образцов и регистрации спектров, а также модели спектрометров.
Идентификация неизвестного вещества по его ИК-спектру заключается в сопоставлении его спектра с эталонным, приведенным в атласе. Учитывая, что колебательные спектры, зарегистрированные на различных спектрометрах или в различных условиях, могут отличаться между собой, важнейшим условие сравнения спектров является стандартизация условий их регистрации. Информационно-поисковые системы, созданные на базе современной электронно-вычислительной техники, помогают отыскать нужный спектр в атласе.
Совпадение спектральной кривой исследуемого вещества со спектральной кривой эталона свидетельствует об идентичности двух веществ. Отсутствие в спектре исследуемого вещества полос, наблюдаемых в спектре образца сравнения, однозначно указывает на то, что эти вещества различны. Присутствие в спектре исследуемого вещества большего числа полос по сравнению со спектром эталона может быть объяснено как загрязнением исследуемого вещества, так и различием обоих веществ.
Таблица 13.3. Характеристические частоты функциональных групп
в органических соединениях
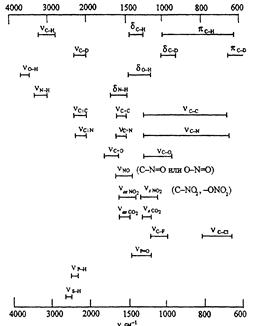
При идентификации молекул органических веществ особое внимание уделяют области спектра 1300-600 см-1. В эту область попадают полосы, отвечающие колебаниям одинарных связей С–С, C–N, С–О, а также многие деформационные колебания. В результате сильного взаимодействия этих колебаний отнесение полос к отдельным связям невозможно, однако весь набор полос в этой области спектра является характеристикой ядерного остова (скелета) молекулы в целом. Эту область называют областью отпечатков пальцев. По колебательным спектрам в этой области можно идентифицировать даже изомеры (табл. 13.3). По этим спектрам (область «отпечатков пальцев»), используемым для идентификации соединений, обычно проводят качественный анализ смесей органических соединений и индивидуальных веществ. Однако надежность такой идентификации при анализе смесей не очень велика, так как многие функциональные группы характерны для разных веществ, что приводит к перекрыванию полос поглощения, затрудняющему идентификацию.
Это хорошо видно из рис. 13.9, на котором изображены ИК-спектры продуктов деструкции сополимеров стирола. Эти токсичные соединения (метилметакрилат, циановодородная кислота, акрилонитрил, аммиак, стирол, формальдегид и др.) загрязняют воздух рабочей зоны и создают неблагоприятные условия для рабочих цеха. Однако, как и в случае УФ-спектров, эти ЛОС образуют широкие и перекрывающие пики, а полосы поглощения в ИК-области спектра одинаковы для многих компонентов смеси загрязнений (ароматические углеводороды С6–C8, ацетальдегид, бензальдегид, акрилонитрил, формальдегид и др.).
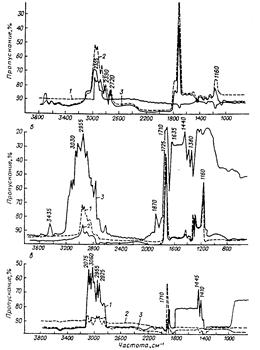
Рис. 13.9. ИК-спектры летучих продуктов деструкции сополимеров стирола – АБС-2501-К (а), МСН (б) и СФН-34 (в): 1 – 150°С; 2 – 225°С; 3 – 250-270°С.
|
|
|
|
Дата добавления: 2014-01-05; Просмотров: 2561; Нарушение авторских прав?; Мы поможем в написании вашей работы!