КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Перенесений гепатит В 18 страница
|
|
|
|
Критерії якості лікування [15, 16, 31]:
1. Відсутність або зворотний розвиток клінічних ознак загострення.
2. Нормалізація або зниження показників запалення і нормалізація імун-
ного статусу.
3. Нормальна або незначно знижена функція нирок.
4. Поліпшення і стабілізація гематологічних порушень.
Лікування резистентної форми СЧВ включає в якості патогенетичної терапії лікарські препарати інших класів — противірусні, андрогеноподібні, простагландини тощо.
Прогноз нині значно покращився: 5-річне виживання досягло 86-95%, 10-річне — 70-80%), а 20-літнє — 60-70%. Однак смертність при СЧВ у 3 рази вища, ніж у популяції.
До факторів несприятливого прогнозу з урахуванням рекомендацій доказової медицини відноситься ураження нирок (особливо дифузний проліфера-тивний гломерулонефрит), АГ, чоловіча стать, початок захворювання у віці до 20 років, супутній антифосфоліпідний синдром, висока активність захворювання, високі значення індексу ураження, приєднання інфекції, ускладнення медикаментозної терапії [30]. Згідно з рекомендаціями Європейської протиревматичної ліги [2007], прогностичну цінність ознак при СЧВ мають наступні ознаки: шкірна висипка, артрит, серозит, судоми / психоз, тяжка анемія, тромбоцитопенія, сироватковий креатинін, протеїнурія / сечовий осад, С3/С4анти-нДНК, анти-Ro/SSA, антифосфоліпідні антитіла, анти-РНП, МРТ головного мозку, нефробіопсія (рівень доказовості В).
Профілактика СЧВ спрямована на усунення захворювання та його загострень у наступних напрямках [15, 25]:
— диспансерне спостереження та проведення адекватного лікування;
— виключення інсоляції, переохолодження, перевтоми, надмірної фізичної активності;
— утримання від оперативних втручань, введення вакцин, сироваток, проведення щеплень (окрім життєво необхідних);
|
|
|
— застосування гормональних препаратів тривало, в строго призначеній дозі;
— не слід приймати пероральні контрацептиви з великим вмістом естрогену, оскільки вони можуть викликати загострення СЧВ.
6.7. ВУЗЛИКОВИЙ ПОЛІАРТЕРІЇТ
Визначення. Вузликовий пол'шртеріїт — це системний некротизуючий васкуліт з переважним ураженнями артерій м'язового типу середнього та дрібного калібру та вторинними змінами в органах і системах.
Етіологія захворювання остаточно не з'ясована. У розвитку вузликового поліартеріїту певне значення надають вірусу гепатиту В. Встановлено, що факторами, які сприяють розвитку захворювання, є перенесені інфекції, введення лікарських препаратів, вакцин, сироваток, інтоксикації, надмірні інсоляції та переохолодження.
Патогенез захворювання зводиться до гіперергічної реакції організму у відповідь на етіологічні фактори. Відбувається розвиток аутоімунних реакцій, утворення комплексів антиген-антитіло, котрі відкладаються в судинній стінці і призводять до збільшення синтезу цитокінів і виникнення запалення.
Імунні комплекси призводять до ураження судин і утворення хемоток-сичних речовин, які привертають у вогнище ураження нейтрофіли. Утворені імунні комплекси у великій кількості виділяють протеолітичні ферменти, які пошкоджують судинну стінку. Слід підкреслити, що в патогенезі захворювання має значення здатність нейтрофілів прилипати до ендотелію та синтезувати за присутності комплементу активовані кисневі радикали, що посилюють пошкодження клітинних мембран судин. Поряд з цим ендотелій судин збільшує виділення факторів, які сприяють підвищенню згортання крові та розвитку тромбоутворень.
Клінічна класифікація (Асоціація ревматологів України, 2004):
Перебіг: гострий, підгострий, хронічний
|
|
|
Активність процесу: відсутня (0), мінімальна (1), помірна (II), висока (III). Стадія: початкова, розгорнута, термінальна. Клініко-морфологічна характеристика ураження:
• шкіра — судинна папуло-петехіальна пурпура, бульозні, везикулярні висипання, ліведо, некротичні зміни шкіри, дигітальний некроз фаланг пальців, рідко — підшкірні вузлики;
• кістково-м'язова система — суглобово-м'язовий синдром (артрит, артралгії, міастенічний синдром з міалгіями;
• периферична НС — асиметрична полінейропатія (ураження черепно-мозкових нервів, ліктьового нерва тощо);
• ЦНС — інфаркт мозку, геморагічний інсульт, психози;
• нирки — судинний тип патології, іноді інфаркти, ХНН, рідко — гломерулонефрит;
• легені — васкуліт легеневий, інтерстиціальна пневмонія, плеврит, інфаркт міокарда;
• система кровообігу — коронарний ІМ, АГ;
• ШКТ — панкреатит, холецистити, кровотечі, інфаркт печінки, гематоми;
• ендокринна система та очі — орхіт, епідидиміт, кон'юнктивіт, ірит, увеіт.
Варіанти перебігу вузликового поліартеріїту: доброякісний, поволі прогресуючий, рецидивуючий, швидко прогресуючий, гострий (злоякісний, блискавичний).
Приклади формулювання діагнозу:
Вузликовий поліартеріїт, класичний варіант, повільно прогресуючий перебіг, активна фаза, активність І з ураженням нирок (нефропатія з сечовим синдромом, ХНН І ступеня), серця (коронарит), СН І стадія, ФК II.
Клініка. Виділяють великі та малі діагностичні критерії вузликового полі-артеріїту [Семенкова Е. Н., 1988].
Великі критерії:
1. Ураження нирок.
2. Коронарит.
3. Абдомінальний синдром.
4. Поліневрит.
5. Бронхіальна астма з еозинофілією.
Малі критерії:
1. Лихоманка.
2. Зниження маси тіла.
3. Міалгічний синдром.
| Діагностичні критерії вузликового поліартеріїту [Американська колегія ревматологів, АСк, 1990] |

Діагноз достовірний при наявності трьох великих і двох малих критеріїв. Інші діагностичні критерії наведені в табл. 6.40.
Лікування захворювання розглянуто в алгоритмі 6.13.
Алгоритм 6ЛЗ. Стандарти лікування вузликового полі артеріїту [6] Крок 1
Основне лікування здійснюється: гормональними (глюкокортикосте-роїди) та не гормональними (цитостатики) препаратами. Глюкокортикоїд преднізолон призначають спочатку в дозі 1-2 мг/кг/добу. Згідно з рекомендаціями Е. Н. Семенкової [1988], в гостру фазу хвороби призначаємо преднізолон у дозі 30-40 мг добу, хворим з астматичним варіантом — 40-60 мг/добу, поєднуючи пероральне введення препарату з парентеральним
|
|
|
Імунодепресант азатіоприн (імуран) призначаємо в дозі 1-2 мг/кг, цикло-фосфамід (циклофосфан) — у дозі 1-2 мг/кг/добу. При блискавичній формі захворювання призначають пульс-терапію: 1000 мг метилпреднізолону в/в З дні підряд і один день приєднують 1000 мг циклофосфану в/в
Крок 2
Застосовують НПЗПта амінохінолінові препарати упродовж активного періоду, які діють протизапально, знеболююче:
—диклофенак — 75-150 мг/добу;
—німесулід — 100-200 мг/добу;
—моваліс — 7,5 мг/добу;
—кеторолаку трометамін (кеторол) 30 мг/добу
—гідроксихлорохін — 0,2 г/добу
Крок З
Застосовують антикоагулянти, антиагреганти та ангіопротекто-
ри при гіперкоагуляції, гіперагрегації тромбоцитів, порушеннях мікроцир-куляції, ДВЗ-синдромі:
—гепарин — 20 тис ОД/добу 1-2 міс;
—фраксипарин — 0,3-0,6 мл/добу 1-2 міс;
—дипіридамол (курантил) — 225-300 мг/добу 3 міс;
—трентал (пентоксифілін, агапурин) — 600-1200, а потім 300 мг/добу
Крок 4
Корекцію AT проводять за допомогою інгібіторів АПФ, діуретиків, БАБ, АК та блокаторів рецепторів ангіотензину II
Крок 5
Застосовують еферентну терапію при відсутності ефекту від імуно-депресантів і протипоказань до них:
—гемосорбція — на курс 3-5 процедур;
—плазмаферез — на курс 3-5 процедур;
— імуносрбція — на курс 3-5 процедур ____________________________________
Наводимо тактику лікування вузликового поліартеріїту в табл. 6.41.
Таблиця 6.41
Тактика лікування хворих з важкими формами вузликового поліартеріїту [Savage С. et аі., 1997]
| Етап | Метод лікування | ||
| Ескалаційна терапія | Активний важкий перебіг із підвищеним рівнем креатиніну > 500 мкмоль/л або з легеневими геморагіями — 7-10 процедур плазмаферезу впродовж 14 днів (видалення плазми в об'ємі 60 мл/кг із заміщенням її відповідним об'ємом 4,5-5%-ного розчину альбуміну) або пульс-терапія метилпреднізолоном (15 мг/кг/добу) впродовж 3-х днів) | ||
| Індукційна терапія 4-6 міс | Цитостатик | Глюкокортикоїд | |
|
|
|
Критерії ефективності лікування: позитивна динаміка клініки, нормалізація лабораторних та імунологічних показників активності запального процесу (лейкоцити, імуноглобуліни, антинуклеарний фактор, ЦІК, морфологічні зміни у судинах).
Прогноз. Терапія вузликового поліартеріїту комбінацією глюкокортико-їдів та цитостатиків призводить до 5-річного виживання в 60-80% випадків. У більшості випадків безпосередньою причиною смерті є серцево-судинні катастрофи. До несприятливих прогностичних факторів належать: вік старше 50 років, ураження нирок, серця, органів травлення, ЦНС.
Профілактика. Первинна профілактика — це запобігання виникненню етіологічних факторів захворювання. Вторинна профілактика вузликового поліартеріїту проводиться із застосуванням імунодепресантів, НПЗП, анти-агрегантів, гіпотензивних засобів в оптимальних підтримуючих дозах тривалий час для уникнення рецидивів захворювання.
6.8. ДЕРМАТОПОЛІМІОЗИТ
Визначення. Дерматополіміозшп — це прогресуюче системне запальне захворювання з ураженням шкіри та поперечносмугастої мускулатури з порушенням рухливої функції та різних процесів у стравоході, легенях і, рідше, серці.
Етіологія та патогенез досі невідомі. У розвитку цього захворювання мають значення наступні патогенетичні фактори:
1) інфекційно-токсична гіпотеза, в якій велика роль надається вірусній інфекції (вірусіам Коксакі, В, А9 тощо), перенесеному грипі, краснусі, оперізуючому лишаю;
2) зв'язок захворювання із злоякісними новоутвореннями (паранеопластич-ний дерматоміозит) як імунопатологічна реакція внаслідок спільності антигенів пухлини і м'язової тканини;
3) імунопатологічна теорія, згідно з якою виявлена сенсибілізація лімфоцитів до антигенів м'язової тканини (в біоптаті м'язів — лімфоплазматичні інфільтрати, як при класичних аутоімунних захворюваннях);
4) генетичні фактори (визначаються НЬА антигени).
Однак основним патогенетичним фактором дерматоміозиту (поліміозиту) є аутоімунний механізм з утворенням аутоантитіл, направлених проти цитоплазматичних білків і рибонуклеїнових кислот, що входять до складу м'язової тканини. Розвитку аутоімунних механізмів сприяє дисбаланс Т- і В-лімфоцитів і зниження Т-супресорної функції. Велике патогенетичне значення має цитотоксичний ефект Т-лімфоцитів проти клітин м'язової тканини.
| Класифікація дерматополіміозиту [Тареев Е. М., Гусєва Н. Г., 1965] |

Класифікація дерматополіміозиту (поліміозиту) наведена в табл. 6.42.
| Д. Ступінь активності | І, II, III |
| Е | Основні клінічні ознаки (синдроми) |
Класифікація дерматоміозиту за С. M. Barson [1966] у модифікації A. Bohan, J. В. Peter [1975]:
1. Первинний ідіопатичний поліміозит.
2. Первинний ідіопатичний дерматоміозит.
3. Дерматоміозит (поліміозит), що поєднується з пухлинами.
4. Дерматоміозит (поліміозит), що поєднується з васкулітом.
5. Поєднання дерматоміозиту (поліміозиту) з дифузними захворюваннями
сполучної тканини.
Рубрикація захворювання згідно з МКХ-10:
M 33 Дерматополіміозит.
M 33.0 Ювенільний поліміозит.
M 33.1 Інший дерматоміозит.
M 33.2 Поліміозит.
M 33.9 Дерматополіміозит неуточнений. Приклади формулювання діагнозу:
1. Первинний ідіопатичний дерматоміозит, гострий перебіг, маніфестна стадія з ураженням м'язів нижніх кінцівок, легень. ЛН II ст.
2. Рак лівої молочної залози. Паранеопластичний дерматоміозит, підго-стрий період з ураженням шкіри, м'язів, судин.
| Діагностичні критерії дерматоміозиту (поліміозиту) (Американська ревматологічна асоціація) |

Клініка. Запропоновані різні діагностичні критерії захворювання (табл. 6.43).
Діагноз дерматоміозиту достовірний:
— при наявності першого основного критерію;
— при наявності двох інших із основних критеріїв;
— при наявності одного основного критерію та двох допоміжних критеріїв. Діагноз поліміозиту достовірний при наявності чотирьох критеріїв без висипу.
Для діагностики дерматоміозиту широко використовують діагностичні критерії Тапітоіо еі аі. [1995]:
1. Зміни шкіри:
— геліотропний висип (пурпурно-червона еритема з набряком верхніх повік);
— симптом Готтона — атрофічна еритема, що злущується на тильній стороні суглобів пальців кисті;
— еритема на розгинальній стороні суглобів кінцівок трохи виступає над поверхнею шкіри, з помірним лущенням, блідо-фіолетова еритема над ліктьовими та колінними суглобами.
2. Слабкість проксимальних м'язів верхніх чи нижніх кінцівок або тулуба.
3. Підвищений рівень сироваткової креатинфосфокінази чи альдолази.
4. Біль у м'язах при натисканні чи спонтанний.
5. Зміни електроміограми (короткі багатофазові потенціали, фібриляції та
псевдоміотонічні розряди).
6. Позитивний тест на антитіло-1 (гістатидил — ИША система) антитіла.
7. Недеструктивний артрит чи артралгії.
8. Ознаки системного запалення (лихоманка > 37° С, ШОЕ > 20 мм/год).
9. Гістологічні ознаки міозиту (запальна інфільтрація скелетних м'язів з фо-
кальною чи поширеною дегенерацією або некрозом).
При наявності хоч би одного шкірного і як мінімум чотирьох критеріїв (від 2 до 9) діагноз дерматополіміозиту досить імовірний.
За даними літератури, спостерігаються наступні зміни лабораторних і гістологічних показників:
• Неспецифічні зміни:
— підвищення активності м'язових ферментів — КФК;
— підвищення ШОЕ (у 50% хворих).
• Неспецифічні для міозиту аутоантитіла:
— антинуклеарні антитіла (АНА) (50-80%>);
— антитіла до РНК (змішані захворювання сполучної тканини і оуег1арріп§-Бупатоте);
— антитіла до РМ-БсІ (поєднання поліміозиту із склеродермією).
• Аутоантитіла, специфічні для міозиту (антитіла до синтетази — анти-тіло-1; анти — БЬІР — антитіла; анти — Мі — 2 — антитіла).
• Біопсія м'язів:
— лімфоплазмоцитарна інфільтрація м'язових волокон і навколо дрібних судин;
— набряк м'язових волокон з втратою посмугованості, фрагментація та дегенерація аж до некрозу міофібрил, фагоцитоз некротичних мас, а пізніше виявляється заміщення некротичних м'язових волокон фіброзною тканиною.
Лікування. Препаратами вибору в лікуванні дерматоміозиту (поліміозиту) є глюкокортикостероїди.
Алгоритм 6.14. Стандарти лікування дерматополіміозиту [6]
Крок 1
Протизапальна терапія:
1. Призначають глюкокортикостероїд преднізолон у дозі 1-1,5 мг/кг/добу в два прийоми до настання ремісії захворювання (протягом 1-3 міс), потім дозу поступово зменшують під контролем клінічних показників. При гострому перебігу доза преднізолону коливається від 80 до 120 мг/добу, підгост-рому — 60-80 мг/добу, при хронічному перебігу — 30-40 мг/добу. Протипоказаний тріамцинолон (полькортолон), оскільки він може посилювати м'язову слабкість (викликати міопатичний синдром) (!). Пульс-терапію проводять при дерматополіміозиті у юнацькому віці у хворих з важким перебігом захворювання
2. Амінохінолінові препарати хлорохін (делагіл) по 0,25 г/добу, гідрокси-хлорохін (плаквеніл) по 0,2 г/добу (протягом 2 років і більше) менш ефективні, їх призначають при слабоактивному процесі
3. НПЗП (диклофенак, індометацин, німесулід, моваліс) застосовують в оптимальних дозах при домінуючому больовому та суглобовому синдромах
Крок 2
Імунодепресанти застосовують при загрозливих для життя станах і неефективності стероїдів. Найчастіше призначають циклоспорин А (сан-димун) у дозі 5 мг/кг/добу, метотрексат — 10-30 мг/тиж. чи азатіоприн — 1-2 мг/кг/добу в/в чи перорально. Оптимальні дози препаратів застосовують упродовж 4-8 тиж, а потім доза знижується до підтримуючої, яку можна призначати роками
При недостатній ефективності рекомендується в/в введення імуноглобу-ліну (1 г/кг 2 дні чи 0,5 мг/кг 4 дні щомісяця впродовж 4 міс)
Крок З
Препарати, що поліпшують метаболізм в уражених м'язах
Застосовують анаболічні гормони для посилення м'язової сили та збільшення об'єму м'язів:
— нероболіл, силаболіл, дураболіл тощо по 1 ін'єкції в/м 1 раз в 10 днів, на курс 3-4 ін'єкції, курси проводити кожний квартал. Показаний рибоксин по 600 мг/добу впродовж 2 міс;
— вітаміни, особливо групи В
Крок 4
У разі розвитку кальцинозу застосовують комплексони, що утворюють розчинні з'єднання з іонами кальцію і сприяють виділенню їх з сечею. Застосовують двонатрієву сіль етілендіамінтетраоцитової кислоти (№2 ЕДТА) в дозі 250 мг щоденно в/в упродовж 5 днів з 5-денними перервами (на курс 15 процедур). Такі курси повторюють 3 рази на рік
Крок 5
Екстракорпоральні методи терапії: плазмаферез (по 5 процедур через день), гемосорбція, імуносорбція
Крок 6
При хронічному перебігу захворювання і відсутності симптомів загострення призначають фізіотерапію: електрофорез з гіалуронідазою, лідазою, парафінові аплікації тощо
Критерії якості лікування:
1. Зниження або відсутність м'язової слабкості чи болю в м'язах.
2. Нормалізація активності ферментів (креатинфосфокінази, альдолази, АсАТ, АлАТ).
3. Нормалізація показників гострофазового запалення (фібриноген, серо-мукоїд, дифеніламінова проба, СРБ, ШОЕ, глобуліни).
4. Нормалізація або поліпшення даних біопсії м'язів, а також даних електроміографії.
Прогноз захворювання серйозний.
Профілактика дерматоміозиту (поліміозиту), яка спрямована на запобігання розвитку загострень, в основному вторинна. З цією метою тривалий час застосовують преднізолон, метотрексат, делагіл тощо. Таким пацієнтам необхідно уникати травм, інфекцій, операцій, щеплень, переохолодження, лікарських препаратів, на які виникають виражені побічні реакції. Жінкам, хворим на дерматополіміозит, слід уникати вагітності.
6.9. СИСТЕМНА СКЛЕРОДЕРМІЯ
Розповсюдженість захворювання складає в середньому 240-290 випадків на 1 млн населення. Частіше хворіють особи негроїдної, ніж європеоїдної раси. Співвідношення захворювань жінок і чоловіків складає 7:1. Термін «склеродермія», або «твердошкір'я» запропонував Сіпітас у 1847 р. [4].
Визначення. Системна склеродермія — це дифузне захворювання сполучної тканини, що характеризується прогресуючим фіброзом та судинною патологією за типом облітеруючої мікроангіопатії, що обумовлює розвиток генералі-зованого синдрому Рейно, індуративних змін шкіри, ураження опорно-рухового апарату та внутрішніх органів [22].
Етіологія захворювання вивчена недостатньо. Припускають вірусне та спадкове походження захворювання.
Патогенез. Визначають наступні основні ланки патогенезу системної склеродермії:
1) надмірне колагенове утворення в результаті порушення функціонування фібробластів; підвищення біосинтезу колагену і та ii типу, глікозаміногліканів, протеогліканів, фібринектину, глікопротеїнів; колагенових волокон та глікоза-мінів, що мають антигенні властивості і призводять до синтезу антитіл за рахунок аутоімунних реакцій;
2) різко наступають судинні, позасудинні та внутрішньосудинні мікроцирку-ляторні зміни, що призводять до метаболічних порушень;
3) відбувається підвищення активності міофібробластів судинної стінки, розвивається деструкція ендотелію, проліферація гладеньком'язових клітин і колагену;
4) збільшується синтез тромбоксану, ендотеліну, зменшується утворення оксиду азоту, що сприяє виникненню спазму судин;
5) виникає гіперкоагуляція, підвищується агрегація тромбоцитів і еритроцитів, розвивається мікротромбоз і генералізований синдром Рейно.
Класифікація. В практичній роботі використовується робоча класифікація системної склеродермії [Н. Г. Гусєва, 1975]:
1. Характер перебігу: гострий, підгострий, хронічний.
2. Стадія розвитку:
i стадія — початкова (синдром Рейно, поліартралгії чи артрит, рідше — шкірні, вісцеральні чи загальні прояви).
ii стадія — генералізована (полісиндромність та полісимптомність клініч-
ної картини).
iii стадія — термінальна (важкість склеротичних, дистрофічних і судинно-некро-
тичних змін у різних системах і органах, нерідко з недостатністю їх функцій).
3. Ступені активності:
I ступінь — мінімальний;
II ступінь — помірний;
III ступінь — високий.
4. Клініко-морфологічна характеристика.
Ураження шкіри та периферичних судин: «щільний» набряк, індура-ція, атрофія, гіперпігментація, телеангіоектазії, синдром Рейно, вогнищеве ураження.
Ураження опорно-рухового апарату: артралгії, поліартрит, кальциноз, ос-теоліз.
Ураження серця: міокардит, кардіосклероз, вада серця (здебільшого міт-ральна недостатність).
Ураження легень: інтерстиціальна пневмонія, пневмосклероз, адгезивний плеврит.
Ураження травного тракту: езофагіт, дуоденіт, коліт, спруподібний синдром.
Ураження нирок: справжня склеродермічна нирка, хронічний дифузний нефрит, вогнищевий нефрит.
Ураження нервової системи: поліневрит, нейропсихічні розлади, вегетативні порушення.
Рубрикація згідно з МКХ-10
М 34.0 Прогресуючий системний склероз М34.1 Синдром CREST
М 34.2 Системний склероз, спричинений лікарськими препаратами і хімічними речовинами
М 34.8 Системний склероз неуточнений.
Виділяють такі клінічні форми системної склеродермії [LeRoy Е. С. et al., 1988; Black С. ML, 1995]:
1. Дифузна форма:
— генералізовані ураження шкіри кінцівок, лиця, тулуба протягом року; синдром Рейно виникає одночасно чи після ураження шкіри;
— ранній розвиток вісцеральної патології (легень, серця, нирок, органів травлення);
— значна редукція капілярів нігтя з формулюванням аваскулярних ділянок;
— виявлення антитіл до топоізомерази-1 (Scl-70).
2. Лімітована форма:
— тривалий період ізольованого феномену Рейно;
— ураження шкіри обмежено зоною обличчя, п'ястей / стоп;
— пізній розвиток легеневої гіпертензії, ураження органів травлення, теле-ангіоектазії, кальциноз (CREST-синдром);
— виявлення антицентромірних антитіл;
— розширення капілярів нігтя без виражених аваскулярних ділянок. 3. Склеродермія без склеродерми:
— немає ущільнення шкіри;
— феномен Рейно;
— ознаки легеневого фіброзу, гострої склеродермічної нирки, ураження серця, органів травлення;
— виявлення антинуклеарних антитіл (Sel -70, АСА, нуклеолярних).
Перехресна форма:
— характерні поєднання клінічних ознак системної склеродермії чи декількох системних захворювань сполучної тканини.
Ювенільна форма:
— початок захворювання до 16 років;
— ураження шкіри по типу вогнищевої чи лінійної (гемі-форма) склеродермії;
— схильність до утворення контрактур, можливої аномалії розвитку кінцівок;
— помірна вісцеральна патологія, виявлена за допомогою інструментальних методів.
Передсклеродермія:
— ізольований феномен Рейно у поєднанні з капіляроскопічними змінами чи імунологічними порушеннями, характерними для системної склеродермії.
Індуцирована склеродермія:
— розповсюджене, частіше дифузне ураження шкіри (індурація), яка інколи поєднується з судинною патологією.
Приклади формулювання діагнозу:
1. Системна склеродермія, гострий перебіг, стадія II, активна фаза, активність II ст., з ураженням шкіри, суглобів, судин (синдром Рейно), поліартрит, ФНС І ст.
Клініка. Діагностичні критерії Американської ревматологічної асоціації [1980]
Великі критерії: склеродермічні ураження шкіри, обличчя, шиї, тулуба (проксимальна склеродермія); ущільнення та індурація шкіри пальців рук та плюснефалангових суглобів.
Малі критерії: склеродактилія, рубці на подушечках пальців, симетричний базальний пневмосклероз.
Діагноз системної склеродермії достовірний при наявності одного великого та двох малих критеріїв.
Дані критерії характеризуються високою специфічністю, однак їх чутливість недостатня.
Критерії діагностики системної склеродермії [Гусєва Н. Г., 1993, 19971 (з доповненнями) Основні критерії:
1. Склеродермічніураження шкіри, що проходять послідовно стадії «щільного» набряку, індурації та атрофії з переважною локалізацією на обличчі (маскоподібність) і в ділянці п'ястей (склеродактилія); можливе тотальне ураження. Шкірний синдром поєднується з пігментацією.
2. Симптом «кисету» — зменшення ротової апертури, потоншення червоної кайми губ, навколо яких формуються радіальні складки (рис. 6.8).
3. Виразкові ураження шкіри на ділянках ліктьових та колінних суглобів, у ділянці щиколоток та п'яток.
4. Суха гангрена — некроз шкіри та підшкірних м'яких тканин розпочинається з дистальних фаланг пальців і може розповсюджуватись на середні фаланги з подальшою демаркацією та самоампутацією.
5. Дигітальнірубчики — точечні ділянки атрофії шкіри дистальних фаланг пальців п'ястей («пацюковий» укус).
6. Синдром Рейно.
7. Суглобово-м'язовіш синдром з розвитком стійких контрактур, в основі якого — ревматоїдоподібний артрит, періартикулярні зміни та фіброзуючий міозит.
8. Остеоліз нігтьових, а інколи середніх і основних фаланг пальців рук, рідше — ніг, що супроводжується вкороченням і деформацією пальців (рис. 6.8).
9. Синдром Тібьєржа-Веііссенбаха — відкладення солей кальцію переважно в ділянці пальців рук і періартикулярно — навколо ліктьових, плечових і кульшових суглобів, у підшкірній клітковині, інколи по ходу фасцій та сухожильних м'язів.
10. Ураження харчового тракту (склеродермічний езофагіт з дисфагією, дилатація стравоходу, гастрит, дуоденіт, порушення моторики кишечника з розвитком кишкової непрохідності, синдрому мальабсорбції).
11. Ураження слизових оболонок: потовщення чи вкорочення вуздечки язика.
12. Ураження серця по типу первинного великовогнищевого кардіосклерозу.
13. Ураження легень по типу базального пневмосклерозу, кистозної легені (на рентгенограмі — «медові соти») (рис. 6.8).
14. Справжня склеродермічпа нирка, яка діагностується клінічно на основі раптового підвищення АТ і розвитку гострої ниркової недостатності.
15. Наявність специфічних антинуклеарних антитіл (анти-8с1-70 та анти-центромірних антитіл).
16. Капіляроскопічні ознаки (за даними капіляроскопії).
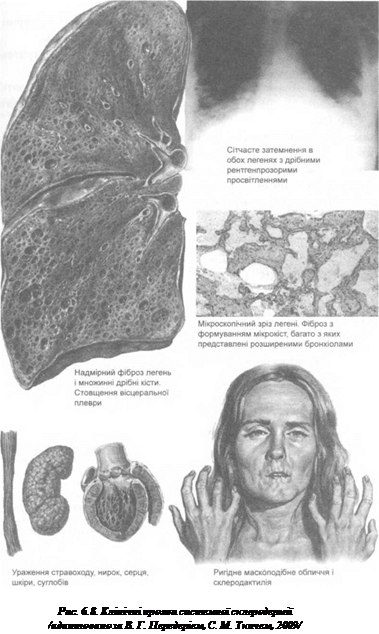
Допоміжні малі критерії:
1. Периферичні: гіперпігментація шкіри, телеангіоектазії, трофічні порушення, синдром Шегрена, поліартралгії, поліміалгії, поліміозит.
2. Вісцеральні: полісерозит, хронічна нефропатія, поліневрит, тригемініт.
3. Загальні: втрата маси тіла (більше 10 кг).
4. Лабораторні: збільшення ШОЕ (більше 30 мм/год), гіперпротеїнемія (вміст білка більше 85 г/л), гіпергамаглобулінемія (більше 23%), антитіла до ДНК чи АНФ, РФ.
| Клінічне специфічне ураження органів і систем при системній склеродермії [36] |

Наявність будь-яких трьох основних критеріїв чи одного із основних, якщо ним є склеродермічне ураження шкіри, остеоліз нігтьових фаланг або характерні ураження харчового тракту, у поєднанні з трьома і більше допоміжними ознаками, є достатніми для достовірного діагнозу системної склеродермії. При меншій кількості симптомів виставляють тільки «можливий» діагноз захворювання.
| Органи травлення | Надлишковий ріст мікрофлори | Анемія |
| Виразки та стриктури стравоходу Баретта | ||
| Васкулярна ектазія антраль-ного відділу шлунка | Анемія, кровотеча | |
| Гастроезофагеальний ре-флюкс | Хронічний кашель, дисфагія, неприємний запах із рота, фарингіт | |
| Мальабсорбція | Схуднення, діарея | |
| Псевдонепрохідність | Симптоматика непрохідності кишечника | |
| Сечостатева система | Статева дисфункція | Імпотенція, дискомфорт при статевому акті |
| Гостра нефропатія | АГ, офтальмоскопічні зміни, наявність цистоцитів у мазку периферичної крові | |
| Шкіра | Кальциноз | Відкладання кальцію в сухожиллях і розгиначах пальців |
| Свербіж | Розчухування | |
| Телеангіоектазії | Судинні утворення, які бліднуть при пальпації | |
| Ущільнення шкіри | Зменшення розміру ротового отвору, склеродактилія, натягнута шкіра | |
| Опорнорухова система | Згинальні контрактури | Неможливість зведення п'ясті внаслідок неповного розгинання пальців |
| Атрофія м'язів | Слабкість | |
| Набряк п'ясті | Дифузний набряк п'ясті, неможливість згинання пальців у кулак | |
| Шум тертя сухожилів | Визначається при пальпації або аускультації під час активного розгинання пальців, колін, стоп тощо |
Залежно від швидкості прогресування системної склеродермії розрізняють гострий, підгострий і хронічний перебіг.
|
|
|
|
|
Дата добавления: 2017-01-14; Просмотров: 268; Нарушение авторских прав?; Мы поможем в написании вашей работы!