КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Броматометрия
|
|
|
|
Л
+ + 4
Гравиметрический……………Измерение массы определяемого вещества или его
Титриметрический……………Измерение объема израсходованного на реакцию
Знаки тригонометрических функций в зависимости от координатной четверти
Базовые тригонометрические формулы
Решение тригонометрических уравнений сводится к приведению их к простейшему виду.
Тригонометрические уравнения
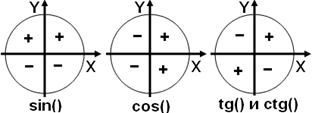
Уравнения, содержащие знак модуля
Самый распространённый метод решения уравнений с модулем – раскрытие модуля согласно определению:
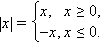
Разобъем числовую ось точками, в которых обращаются в нуль выражения, стоящие под знаком модуля.
Выбирая на этих промежутках контрольные точки, проверяем знак выражения, стоящего под знаком модуля.
Для каждого интервала согласно определению модуля решаем свое уравнение.
Простейший вид приведен ниже в таблице.







раствора реактива точно известной концентрации.
составных частей, выделяемых в виде
соответствующих соединений.
Важнейшими характеристиками методов анализа является их чувствительность и точность. Чувствительностью метода анализа называют наименьшее количество вещества, которое можно достоверно определить данным методом. Точностью анализа называют относительную ошибку определения, которая представляет собой отношение разности найденного (х1) и истинного (х) содержания вещества к истинному содержанию вещества и находят по формуле:
Отн. ош.= (х1–х)/ х, для выражения в процентах умножают на 100. За истинное содержание принимают среднеарифметическое содержание вещества, найденное при анализе пробы в 5 -7 определениях.
Метод Чувствительность, моль/л Точность,%
Титриметрический 10-4 0,2
Гравиметрический 10-5 0,05
Весовым (гравиметрическим) анализом называют метод количественного анализа, при котором количественный состав анализируемого вещества устанавливают на основании измерений масс, путем точного взвешивания массы устойчивого конечного вещества известного состава, в которое полностью переведен данный определяемый компонент. Например, гравиметрическое определение серной к-ты в водном растворе осуществляется с помощью водного раствора соли бария: ВаС12 + Н2SО4→ ВаSО4↓ +2 НСl. Осаждение проводят в таких условиях, в которых практически весь сульфат-ион переходит в осадок ВаSО4 с наибольшей полнотой – количественно, с минимальными потерями, вследствие незначительной, но все же имеющейся растворимости сульфата бария. Далее осадок отделяют от раствора, промывают для удаления растворимых примесей, высушивают, прокаливают, для удаления летучих сорбированных примесей и взвешивают на аналитических весах в виде чистого безводного сульфата бария. А затем рассчитывают массу серной кислоты. Классификация методов гравиметрического анализа. Методы осаждения, отгонки, выделения, термогравиметрические методы (термогравиметрия). Методы осаждения – определяемую составную часть количественно связывают в такое химическое соединение, в виде которого она может быть выделена и взвешена. Состав этого соединения должен быть строго определенным, т.е. точно выражаться химической формулой, и оно не должно содержать каких-либо посторонних примесей. Соединение, в виде которого определяемую составную часть взвешивают, называют весовой формой.. Пример, определение Н2SО4(выше), определение массовой доли железа в его растворимых солях, основанное на осаждении железа (111) в форме гидроксида Fе(ОН)3 хН2О с последующим его отделением и прокаливанием до оксида Fе2О3 (весовая форма). Методы отгонки. Определяемый компонент выделяют из анализируемой пробы в виде газообразного вещества и измеряют либо массу отогнанного вещества (прямой метод), либо массу остатка (косвенный метод). Прямой метод широко используется для определения содержания воды в анализируемых веществах путем ее отгонки из взвешенного образца и конденсации, а затем измеряют объем конденсированной воды в приемнике. По плотности пересчитывают объем воды на массу и, зная массу образца и воды, рассчитывают содержание воды в анализируемой пробе. Косвенный метод отгонки широко применяют для определения содержания летучих веществ (включая слабосвязанную воду) по изменению массы образца до и после высушивания до постоянного веса в термостате (в сушильном шкафу) при постоянной температуре. Условия проведения таких испытаний (температура, время сушки) определяются природой образца и конкретно указываются в методических руководствах. Методы выделения основаны на выделении из раствора определяемого компонента путем электролиза на одном из электродов (электрогравиметрический метод). Затем электрод с выделевшимся веществом промывают, высушивают и взвешивают. По увеличению массы электрода с веществом находят массу выделившегося на электроде вещества (сплавы золота, меди переводят в раствор). Термогравиметрические методы не сопровождаются отделением исследуемого вещества, а исследуется сам образец поэтому эти методы условно относят к гравиметрическим методам анализа. Методы основаны на измерении массы анализируемого вещества при его непрерывном нагревании в заданном температурном интервале на специальных приборах – дериватографах. По полученным термогравиграммам при их расшифровке можно определить содержание влаги и других составляющих анализируемого вещества. Основные этапы гравиметрического определения: расчет массы навески анализируемой пробы и объема (или массы) осадителя; взвешивание (взятие) навески образца; растворение навески анализируемого образца; осаждение, т.е. получение осаждаемой формы определяемого компонента; фильтрование (отделение осадка от маточного раствора); промывание осадка; высушивание и (при необходимости) прокаливание осадка до постоянной массы, т. е. получение гравиметрической формы; взвешивание гравиметрической формы; расчет результатов анализа, их статистическая обработка и представление. Каждая из этих операций имеет свои особенности. При расчете оптимальной массы навески анализируемого вещества учитывают возможную массовую долю определяемого компонента в анализируемой пробе и в гравиметрической форме, массу гравиметрической формы, систематическую ошибку взвешивания на аналитических весах (обычно 0,0002), характер получаемого осадка – аморфный, мелкокристаллический, крупнокристаллический. Расчет исходной навески ведут исходя из того, что масса гравиметрической навески должна быть не меньше 0,1 г. В общем случае нижний предел оптимальной массы m исходной навески анализируемого вещества (в граммах) рассчитывают по формуле: m = 100m (ГФ) F/ W(X), где m(ГФ) – масса гравиметрической формы в граммах; F - гравиметрический фактор, фактор пересчета, аналитический множитель); W(X) – массовая доля (в %) определяемого компонента в анализируемом веществе. Гравиметрический фактор F численно равен массе определяемого компонента в граммах, соответствующий одному грамму гравиметрической формы. Гравиметрический фактор рассчитывают по формуле как отношение молярной массы М(Х) определяемого компонента Х к молярной массе гравиметрической формы М(ГФ), умноженное на число n молей определяемого компонента, из которого получается один моль гравиметрической формы: F = n M(X) / M (ГФ). Так, если из 2-х молей Fе С13 6Н2О получается один моль гравиметрической формы Fе2О3, то n = 2. Если из одного моля Ва(NО3)2 получают один моль гравиметрической формы ВаСrО4, то n = 1.
ТЕМА 7
ГРАВИМЕТРИЧЕСКИЙ (ВЕСОВОЙ) МЕТОД АНАЛИЗА
Гравиметрический анализ. Аналитика строго говоря интересует не вес реагирующих в-в, а их массы. (поскольку вес это непостоянный показатель и изменяется с изменением расстояния от тела до центра земли, на экваторе меньше, чем на полюсах, в невесомости равен нулю). Мерой кол-ва материи (в обычных условиях) является масса, за ед. кот-й принят кг, ед-ей веса является ньютон. Т.о. атомный, молекулярный вес – это условные понятия, принятые в химии. Измерение массы при выполнении опред-ий методом весового анализа чаще всего должно быть очень точным и поэтому проводится обычно при помощи весьма точного прибора – аналитических весов. Дать показатели точности при взвешивании на ан. весах. Выделяются 3-и типа весовых определений. 1 – весовые опред-ия при выполнении кот-ых опред-мую составную часть количественно выделяют из анализ-го в-ва и взвешивают. Количественно выделить – значит выделить составную часть анализируемого в-ва настолько полно, насколько это позволяют сделать лабораторные приемы и св-ва данного в-ва. Так опред-ют например сод-ие золы в каменном угле (это очень важный анализ, т. к. сод-ся в угле примеси снижают его теплотворные кач-ва и засоряют топку). Для проведения такого анализа берут навеску угля, т.е. точно взвешивают небольшое кол-во пробы угля и сжигают его в муфельной печи в тигле (тигель доведен до постоянного веса) и навеску прокаливают до тех пор, пока масса оставшейся золы не будет уменьшаться, тигель с остывшей в эксикаторе золой точно взвешивают и опред-ют зольность угля в процентах, исходя из пропорции: навеска – 100%, масса золы – х%, т.е. х= масса золы 100/навеска. Т.о. методы выделения делятся на выделении в-ва путем сжигания, электролиза (далее стр. 3). 2. определяемую составную часть полностью удаляют, а остаток взвешивают. Таким способом обычно определяют сод-ие влаги в исследуемых в-вах. Привести пример и расчет определения влаги. Условия проведения таких испытаний на стр.3 в методе отгонки. Весовые опред-ия первых 2-х типов выполняются довольно просто, в 3-ем случае метод осаждения, когда невозможно, например выделить и взвесить серу из каменного угля применяется метод осаждения (стр.3). В данном примере серу выделить в виде малорастворимого соединения ВаSО4. По кол-ву бария сернокислого легко опред-ть сод-ие серы, содержащейся в навеске угля, т. к. каждому атому серы соответствует одна мол-ла сер-го бария. Вычисления могут быть проведены при помощи соответствующих пропорций. Для гравиметрических определений существует н-ко общих правил: 1. Перед определением какой-либо составной части анализируемого в-ва из навески взятого образца должны быть тщательно удалены другие составные части, ведущие себя в процессе данного анализа также, как и определяемая, или приняты соответствующие меры, не допускающие выделения этих сопутствующих составных частей вместе с определяемой (при опред-ии серы вместе с барием могут выделяться сульфаты второй группы катионов). 2. Следует использовать реактивы соответствующей степени чистоты: ч.д.а. или х.ч, обычно это указ-ся в методичках). 3. Правильно выполнить расчеты, т.к. ошибка в расчете равноценна ошибке в анализе. Средняя проба анализируемого в-ва – это небольшое кол-во этого в-ва, состав которого одинаков с составом всей партии, от которой взята проба. Правила отбора средней пробы зависят от физического состояния продукта (твердое, жидкое, газообразное), структуры (крупнокусковое, мелкокусковое, зернистое, порошкообразное и т. п.), упаковки (банки, мешки, бочки, навалом, насыпом), способом перевозки (автомобили, железнод-ые вагоны, цистерны, баржи и т.д.), а также размером партии. Методы отбора проб: первичная большая из различных мест партии, квартование, разделение на 3-и равные части: на анализ, на повтор и на арбитражное хранение. Все это определяется НТД на данный продукт. Навеска – небольшое, точно взвешенное кол-во анализируемого в-ва, взятое от средней его пробы, кот-ое в процессе анализа количественно подвергается всем необходимым операциям. Обычно это н-ко граммов или десятых грамма. От количества навески зависит точность результата анализа. Об абсолютной и относительной ошибках мы говорили, ясно что чем больше навеска в-ва тем меньше будет относительная ошибка при одинаковой ошибке взвешивания (после 4-го знака после запятой). Но большая проба требует большего кол-ва реактивов, ее обработка требует большего времени (например сжигание, прокаливание). Поэтому при проведении гравиметрических опред-ий ведут предв-ый расчет навески. Для весовых анализов 1 и 2-го типа навеска должна быть такой величины, чтобы выделяемая и удаляемая из навески определяемая часть составляла от 0,01 до 0,1 г. Следовательно, чтобы правильно рассчитать навеску нужно знать хим-ую формулу анализ-го в-ва или приблизительное сод-ие составной части в пробе. Например, какую навеску образца каменного угля нужно взять для определения его зольности, если предпол-ая зольность 10%?. Т. к. золы в образце меньшее кол-во, то берем большую норму, т.е. 0,1г, чтобы навеска не получилась слишком маленькой и рассчитываем по пропорции: 0,1 -10%, х(навеска) – 100 %, получ-ся 1г. В третьем виде для образования осадка следует учитывать физические св-ва (структуру и плотность) в-ва, в виде которого осаждается определяемая составная часть, т.н. осаждаемая форма, которая часто отличается от весовой формы. Так при определении железа осаждаемая форма Fе(ОН)3, а весовая форма – Fе2О3, в случае ВаSО4 и весовая и осаждаемая форма одинаковы, ионы Са осаждаются в виде СаС2О4Н2О, а взвешиваются после прокаливания осадка в виде СаО. Осаждаемая форма оксалат кальция, а весовая форма – окись кальция. Плотность получаемого осадка влияет на дальнейшие стадии его обработки, особенно на промывку – легко осадка будет много в фильтре и его трудно и долго придется промывать и в этом случае предпочтительнее тяжелый осадок. Поэтому в осадительных методах величина навески должна рассчитываться исходя из характера осадка: аморфного (легкого, гидроокиси) – 0,07 – 0,1г; кристаллического легкого (большинство солей) – 0,1 – 0,15г; кристаллического тяжелого – 0,2 – 0,4г; кристаллического очень тяжелого (соли свинца, серебра) до 0,5г. Взятие навески на аналитических весах требует определенных навыков и выполняется по соответствующим правилам, переносят количественно в стакан или колбу смывая водой и растворяя в воде или к-те при нагревании в водяной или песочной бане и приступают к осаждению. Цель осаждения – количественно перевести определяемую составную часть анал-го в-ва в в опред-ое химич-ое соединение. Зная массу выделенного осадка, можно рассчитать сод-ие опред-ой составной части, если осаждение произойдет не количественно, то масса осадка получится меньшей, а след-но и результат анализа будет меньше. И тут есть правило: если осаждаемый ион может быть осажден различными ионами, из них следует выбирать в кач-ве осаждающего тот, который образует осадок, обладающий наименьшей растворимостью. Для расчета кол-ва реактива необходимого для осаждения исходят из правила эквивалентности согласно которому 1г-экв осаждаемой составной части реагирует с 1 г-экв осадителя. На практике осадителя берут в некотором избытке. При осаждении необходимо соблюдать следующие условия: осаждать только из разбавленных р- ов разбавленными растворами осадителя (концентрированные р-ры следует использовать при получении аморфных осадков). Этот прием позволяет медленно и количественно образовываться осадку и и затрудняет все виды соосаждения; осаждать только подогретые р-ры горячими растворами осадителей, что способствует образ-ию крупных кристаллов; осадитель в кол-ве вычисленного объема добавлять медленно, порциями, при постоянном помешивании стекл-ой палочкой; следует добавлять небольшое кол-во коагул-х в-в, желательно улетучив-ся при прокаливании осадка. Дать время для созревания осадка, т.е. для превращения мелких кристаллов в крупные. Аморфные осадки подвергают дальнейшей обработке сразу, без выдержки для созревания, т.к. они ведут себя в р-ре как адсорбенты, т. е. загрязняются. Проверка полноты осаждения добавлением по стенке стакана н-ких капель осадителя и при появлении мути добавляют н-ко мл раствора осадителя и выдерживают некоторое время на водяной бане. Фильтрование и фильтрование с декантацией – сливание отстоявшейся жидкости по стеклянной палочке, стараясь не взмучить р-р. Используют воронки стеклянные или другие, фильтр складывают, раскрывают, вставляют в воронку, расправляют, смачивают дист-ой водой, между стенками воронки и фильтром не должно оставаться пузырьков воздуха. Затем добавляют 50 мл промывной жидкости взмучивают, дают отстояться и вновь декантируют и так трижды, практически большее количество промывок, что обеспечивает лучшее и быстрое фильтрование и частичную промывку. Следует не отводить палочку от воронки или от стакана с осадком и не класть ее на стол, чтобы не потерять ни капли раствора. При фильтровании важно не потерять осадок и не добавить вес за счет фильтров. Поэтому используют беззольные фильтры, а не обычную фильтровальную бумагу. Т. е. фильтры предварительно подготовленные из которых удалена значительная часть минеральных в-в. Масса золы такого фильтра настолько мала, что ею обычно пренебрегают. Обычно масса золы указывается на обложке упаковки фильтров, если она составляет более 0,0002 г, то есть находится в пределах чувствительности аналитических весов, ее отнимают от массы прокаленного осадка. Перед началом фильтрации выбирают фильтр нужной плотности и наиболее подходящего размера. Черная или красная лента – наиболее крупнопористые фильтры и соответственно быстро фильтрующие, используются для фильтрации и отделения аморфных осадков, например, гидроксидов железа, алюминия и др. Белая лента – фильтры средней плотности, применяются для отделения кристаллических осадков. Синяя лента – фильтры мелкопористые используются для отделения мелкокристаллических осадков, сульфата бария, оксалата кальция. Для последующей промывки учитывают не объем фильтруемой жидкости, а массу определяемого осадка. Он должен заполнять не более половины фильтра, чтобы не возникали проблемы с его промывкой. Используются стеклянные фильтрующие тигли с вакуумной системой, для фильтрования кристаллических осадков. Промывка осуществляется порциями промывной жидкости при ее приливании после профильтровывания предыдущей порции, в воду следует добавлять небольшое кол-во осадителя, чтобы избежать потерь. В случае пептизации, в промывную воду добавляют электролиты-коагуляторы, обычно легко летучие в-ва, удаляющиеся при последующем прокаливании осадка – обычно это растворы, содержащие аммоний. Осадок увлекает с собой посторонние в-ва, находящиеся вр-ре, это наз-ся соосаждением и предтавлен 4-мя видами: окклюзией, изоморфное соосаждение,, соосаждение сообразованием химических соединений, образющихсяя между посторонним в-вом и осаждаемым в-вом, соосаждение в р-те поверхностной адсорбции примесей осадком. 1-ый тип – это процесс захвата микрокропримесей внутрь растущих кристаллов осадка и наиболее трудно устраняется. 2-ой – процесс образования «смешанных кристаллов» ионами основного компонента и микропримеси, имеющими близкие радиусы, например, сульфат бария может увлекать кристаллы перманганата калия, т.к. эти в-ва изоморфны, т. е. образуют совместную пространственную кристаллическую решетку. 3 – ий вид происходит между осаждаемым в-вом и присутствующими в р-ре примесями, например, при осаждении ионов бария раствором серной к-ты, то, в присутствии ионов железа образуется комплексная соединения сульвата бария и железа, при этом следует предварительно гидроксидом аммония удалить примеси железа в виде гидроксида железа, отфильтровав последний. Иногда для удаления примесей используют переосаждение, т. е. осадок, содержащий примесит, например, оксалат кальция и примесь оксалат магния, растворяют в соляной к-те, нейтрализуют и вновь осаждают оксалатом аммония, т.к. концентрация магния становится меньше, то он не влияет на осаждение кальция. 4-ый вид устраняется промывкой и горячей водой, т. к. адсорбция сопровождается десорбцией. Последняя промывка сопровождается обтиранием стеклянной палочки кусочком фильтра и он кладется в в общий фильтр. И затем делают пробу на полноту удаления примесей, из фильтрата берут 2-3 мл р-ра и проводят качественную реакцию, например на хлориды и т.д. Фильтрат затем отбрасывают, если он не содержит основного в-ва и совсем прозрачен. Отделение осадка и прокаливание. После промывки на фильтре остается практически чистый осадок и остается только узнать его массу. Т. к. количественно снять осадок с фильтра невозможно, в большинстве случаев вместо этого фильтр подсушивают в сушильном шкафу на воронке при 90 – 1050 С, переносят в предварительно доведенный до постоянного веса фарфоровый, металлический или платиновый тигль и сжигают с предварительным озолением на электроплитке и в течение 20 -30 мин в муфельной печи (в случае, если осадок не взаимодействует с углеродом) или без фильтра, высыпают осадок на глянцевую бумагу и накрывают химическим стаканом или воронкой. Фильтр с остатками осадка сжигают в тигле (постоянный вес), добавляют осадок и прокаливают до постоянного веса. Расчет ведут в 2-ва приема: 1. Сначала узнают, ск. г определяемой составной части содержится в полученном кол-ве весовой формы. Эту величину находят из соотношения молекулярных или атомных весов определяемой составной части и весовой формы. 2. Затем (как и в первых 2-х случаях) узнают, сколько % составляет масса определяемой составной части от массы всей взятой навески. Температура прокаливания может ориентировочно определяться по цвету каления муфельной печи, от темно-красной – 525 до ослепительно-белой 1400-15000 С.
Фактор пересчета или аналитический фактор: отношение молярной массы определяемого в-ва к молярной массе в-ва, находящегося в осадке Ф= М опред-го в-ва / М в-ва, нах-ся в осадке. Фактор пересчета показывает сколько граммов определяемого в-ва содержит1 г осадка. Например Ф = М (Ва) М (Ва О4) = 137,40 /233,40= 0,5887. По готовой формуле можно вычислить содержание элемента в сложном веществе: % = (Ф/)100, масса полученного осадка,; Ф – фактор пересчета, - навеска исследуемого в-ва.
ТЕМА 8
ТИТРИМЕТРИЧЕСКИЕ (ОБЪЕМНЫЕ) МЕТОДЫ АНАЛИЗА
Титриметрический метод основан на точном измерении объемов веществ, вступающих в химическую реакцию. В этом методе используют растворы реактивов точно известной концентрации – титранты. Процесс медленного прибавления титранта к раствору исследуемого вещества называется титрованием. Момент титрования, когда количество прибавленного титранта становится эквивалентным количеству определяемого вещества, называется эквивалентной точкой титрования или точкой эквивалентности. Её находят с помощью индикаторов или по изменению физико-химических характеристик титруемого раствора. Титриметрический анализ отличается быстротой и точностью полученных результатов.
Классификация и характеристика методов титриметрического анализа.
| Метод | Протекающая реакция | Титранты |
Нейтрализация или кислотно- Нейтрализация НС1, NаОН
основное титрование Оксидиметрия, или окислительно- Окисление–восстановление КМnО4, К2Сr2О7, восстановительное титрование I2, Nа2S2О3
Комплексонометрия Комплексообразование Трилон Б (НЭДТУ)
Осаждение Осаждение трудно- АgNО3, КSСN.
растворимых солей
Техника выполнения титриметрического анализа.
Приготовление раствора по точной навеске вещества. Молярные растворы.
Для приготовления 1 л 1 М раствора вещества подсчитывают его молекулярную массу и вычисляют массу навески по формуле: g = ММ1V/1000, где g – навеска вещества, г; М – молекулярная масса, г; М1 – требуемая молярность раствора; V – вместимость мерной колбы, мл. Нормальные растворы. Навеску вычисляют по формуле g = ЭNV / 1000, где Э – эквивалентная масса вещества, г; N –требуемая нормальность раствора; V – вместимость мерной колбы, мл. Установка титра раствора. Для установки титра растворов, приготовленных по точной навеске, применяют 2-ва способа: отдельных навесок и пипетирования. Более точный, но требующий большего времени для выполнения – способ отдельных навесок. Для его выполнения вычисляют навеску установочного вещества по формуле (см. выше). При этом объем V обычно составляет 25 мл. Затем на аналитических весах взвешивают три навески (равные вычисленной массе) с точностью до 0,0001 г. Эти навески переносят в чистые колбы и растворяют в дистиллированной воде (по 0,25 мл) или в отдельных случаях в кислоте. После полного растворения навески растворы титруют раствором титранта, титр которого необходимо установить. Расчет титра раствора титранта (Тт) проводят по формуле Т = Этgу.в./ Эу..в..Vт., где Эт – эквивалентная масса титранта, г; Эу.в.-эквивалентная масса установочного вещества, г; gу.в.-навеска установочного вещества, г; Vт объем титранта, затраченного на титрование навески, мл. В 3-х опытах титр рассчитывают для каждой навески отдельно и по найденным значениям определяют его среднее значение и по полученной величине находят среднее значение нормальности Nпр и коэффициента нормальности К титранта по формулам Nпр= Тт1000 /Эт; К =Nпр / Nтеор., где Nтеор – расчетная нормальность титранта. В способе пипетирования предварительно рассчитывают навеску установочного вещества по формуле g = ЭNV / 1000, приняв V = 250 мл. Навеску установочного вещества взвешивают на аналитических весах с точностью до 0,0001 г, переводят в мерную колбу вместимостью 250 мл, растворяют. Доводят водой до метки водой и тщательно перемешивают. Помещают в колбу для титрования 25 мл раствора, добавляют индикатор и титруют раствором титранта до изменения окраски индикатора. Нормальность рассчитывают по формуле Nпр= Nу.в.25 / Vт, где Nу.в.– нормальность раствора установочного вещества; Vт – объем титранта, мл. Перечень веществ, применяемых для установки титра важнейших титрантов, и их характеристики приводятся в методических инструкциях и в табл. 113 Справочного руководства по химии, А. И. Артеменко. – М., Высшая школа, 2002 г. Установочные вещества необходимо брать в виде препаратов марки «ХЧ» после их перекристаллизации из воды. При установке титра способом пипетирования целесообразно использовать в качестве установочного вещества фиксанал соответствующего реагента. Метод нейтрализации применяется для определения содержания различных кислот, оснований, кислых и гидролизующихся солей. В основе этого метода лежит реакция Н+ + ОН- = Н2О. Кривые титрования метода нейтрализации. В процессе титрования растворов кислот и оснований рН титруемого раствора непрерывно меняется. Сущность метода. В водных растворах это реакция нейтрализации. Н3О++ОН-=2Н2О. Титрантами метода являются растворы сильных кислот и оснований: НС1, Н2ЅО4, NаОН, КОН. Эти вещества не соответствуют требованиям, предъявляемым к стандартным веществам, поэтому концентрацию титрантов устанавливают стандартизацией их растворов. В качестве первичных стандартов чаще всего используют буру Nа2В4О7.10Н2О, безводный карбонат натрия Nа2СО3, дигидрат щавелевой кислоты Н2С2О4.2Н2О. В процессе титрования растворов кислот или оснований рН титруемого раствора непрерывно меняется. График зависимости рН раствора от количества прибавленного титранта наз-ся кривой титрования. Кривые титрования дают возможность проследить изменение рН раствора в различные моменты титрования, изучить влияние температуры и концентрации реагирующих веществ на процесс нейтрализации, установить конец титрования и помогают сделать правильный выбор индикатора. Построение кривой титрования сводится к вычислению рН и концентрации титруемого вещества, соли и избыточного титранта в точках, когда к 100 мл титруемого раствора прибавлено 0; 10; 50; 90; 99; 100; 101; 110; 150; 190; 200 мл титранта. Полученные данные наносят на график, на оси абсцисс которого отложен объем прибавленного титранта, а на оси ординат – соответствующее ему значение рН, причем рН 14 должно находиться в начале координат, а рН 0 – вверху оси ординат. Полученные точки соединяют плавной кривой по лекалу. Рекомендуемый масштаб изображения: по оси х – ось абсцисс -1см соответствует 10 мл, а по оси у – ось ординат – одной 1 рН. Вычисления концентрации компонентов производят по формулам: в точках 5-99 мл с = (100-V/100+V)N; в точке 100 мл с = (V/100 +V)N; в точках 101 -200 мл с = (V-100/100+V)N, где V – объем прибавленного титранта, мл; N -нормальность раствора. В нулевой точке концентрация титруемого раствора задается условиями задачи. рН рассчитывается по формулам: для сильных кислот: рН = рсα (1), т.е. рН равняется отрицательному десятичному логарифму концентрации к-ты; для сильных гидроксидов КОН, NаОН, Са(ОН)2 – рН =14–рсα (2); для слабых кислот – рН =1/2(рКα +рсα) (3); для слабого гидроксида – рН =14-1/2(рКα +рсα)(4); смесь растворов слабой кислоты и ее соли (кислотный буферный р-р) – СН3СООН и СН3СООNа – рН =рКα +рсα – рсb (5); смесь растворов слабого гидроксида и его соли (щелочной буферный р-р) – NН4ОН, NН4С1 – рН =14–рКb – рсb +рсс (6); для солей, образованных слабым гидроксидом и сильной к-ой – СuЅО4, NН4С1 – рН = 7 -1/2 рКb + 1/2рсс (7); для солей, образованных слабой кислотой и сильным гидроксидом – Nа2СО3, КF – рН = 7 -1/2рКα-1/2рсс (8); для солей, образованных слабой к-ой и слабым гидроксидом – рН =7+1/2рКα+1/2рКb(9), где Кα и Кb – константы электролитической диссоциации кислоты и основания; рКα и рКb – отрицательные логарифмы констант электролитической диссоциации кислот и гидроксидов; сс – концентрация соли, рсс – отрицательный логарифм концентрации соли. Первые два показателя находят по табличным данным и подставляют в соответствующие формулы. Пример. Вычислить рН 0,1 М р-ра уксусной к-ты. Уксусная к-та – слабый электролит и расчет производим по формуле рН = ½(рКα+рсα), рКα для уксусной к-ты равно 4,75. Концентрацию к-ты 0,1 М записываем в виде 10-1, тогда рсс = -1g 10-1 = 1. Подставив найденные значения в формулу, получим рН = ½(4,75 + 1) = 2,85.
Элементы титриметрического анализа. Основные понятия термины и формулы.
Если известны нормальности 2-х растворов и их объемы, при которых достигается точка эквивалентности, то можно вычислить количества эквивалента кислоты и щелочи: n(1/2Н2SО4)=С(1/2Н2SО4).V(Н2SО4)/1000, n(NаОН) = С (NаОН). V (NаОН) /1000, где V (Н2SО4) и V(NаОН) выражены в миллилитрах.
Таким образом, при подстановке правых частей 2-х последних уравнений в предыдущее равенство получим очень важное для титриметрического анализа выражение принципа эквивалентности: С(1/2Н2SО4).V(Н2SО4) =С(NаОН).V(NаОН).
В общем виде для любых случаев титрования это соотношение имеет следующее выражение: Сн(Х).V(Х)=Сн(Т).V(Т), где Сн(Х), Сн(Т) – нормальности раствора анализируемого вещества Х и титранта Т, V(Х) и V(Т) – объемы анализируемого вещества и титранта соответственно. Из этого соотношения следует: V(Т)/V(Х)= Сн(Х)/Сн(Т).
Если известен объем анализируемого раствора, то по последнему уравнению можно рассчитать его нормальность: Сн(Х)= Сн(Т).V(Т)/V(Т).
Вычислив по результатам титрования нормальность анализируемого раствора из вышеприведенных уравнений можно определить массу вещества в любом объеме раствора.
При рассмотрении большинства химических реакций удобно пользоваться понятиями эквивалента и фактора эквивалентности.
Эквивалент – это некая реальная или условная частица, которая может присоединять или высвобождать один ион водорода в кислотно-основных реакциях или один электрон в окислительно- восстановительных реакциях.
В другой формулировке эквивалент – это некая реальная или условная частица, которая в реакции обмена или окисления-восстановления взаимодействует с носителем одного элементарного заряда. Например, в реакции Н3РО4+ ОН- =Н2РО4- +Н2О молекула Н3РО4 равна эквиваленту, т. к. она реагирует с одним ионом ОН-. А в реакции Н3РО4+2ОН- =НРО42-+2Н2О молекула Н3РО4 реагирует с 2-мя ионами ОН-, значит, здесь эквивалент фосфорной кислоты равен ½ молекулы Н3РО4.
Фактор эквивалентности ƒэкв(Х) – это число, показывающее, какая доля реальной частицы вещества Х эквивалента равна одному иону водорода в данной кислотно-основной реакции или одному электрону в окислительно-восстановительной реакции. Это величина безразмерная, рассчитывается на основании стехиометрических коэффициентов конкретной реакции.
Фактор эквивалентности часто обозначают отношением 1/ z, где z – суммарный заряд обменивающихся в молекуле ионов для обменных реакций или число электронов, принятых или отданных молекулой (атомом) вещества, для ОВР; z – всегда целое положительное число, а фактор эквивалентности – меньше или равен 1: ƒэкв(Х) = 1/ z <1. Фактор эквивалентности одного и того же вещества может иметь разные значения в разных реакциях. В связи с этим, при использовании терминов «эквивалент» и «фактор эквивалентности» вещества всегда необходимо указывать, к какой конкретной реакции они относятся.
Единицей количества вещества эквивалента является моль. Наряду с молярной массой вещества М(Х) в количественном анализе широко пользуются понятием молярной массы эквивалента вещества Х, которой называется масса одного моля эквивалентов этого вещества. Она равна произведению фактора эквивалентности вещества в реакции на молярную массу вещества Х: Э(Х)=М(ƒэкв(Х)Х)=ƒэкв(Х).М(Х).
Количество вещества (в молях), в котором частицами являются эквиваленты, называется количеством вещества эквивалента (n э(Х)). Очевидно, что n э(Х) = m (Х)/Э((Х). Например, требуется рассчитать количество вещества эквивалента безводной соды в ее навеске массой 5,3 г при проведении реакции с НСl до СО2, в соответствии с уравнением реакции Nа2СО3+2НСl =2NаСl +Н2О +СО2, ƒэкв(Nа2СО3)=1/2. В результате уравнения реакции имеем n (1/2Nа2СО3) = m (Nа2СО3)/ M (1/2Nа2СО3) =5,3/53 =0,1моль.
Т. о. на основании 2-х последних уравнений легко получить выражение для расчета массы вещества, исходя из количества вещества эквивалента и фактора эквивалентности вещества: M (Х) = nэ (Х).Э(Х) = nэ (Х).ƒэкв(Х). M (Х).
В титриметрическом анализе для выражения состава раствора используют молярную концентрацию эквивалента или нормальность, которая равна количеству вещества эквивалента (в молях), содержащегося в одном литре раствора. Она обозначается С(ƒэкв(Х)Х) (иногда Сн или N с указанием значения фактора эквивалентности вещества) и рассчитывается как отношение количества эквивалента растворенного вещества Х к объему раствора в литрах: Сн(Х)=С(ƒэкв(Х)Х) = n э(Х)/V = m (Х)/Э(Х).V).
Выразив в знаменателе молярную массу эквивалента через молярную массу вещества и фактор эквивалентности его в реакции по уравнению (1), получим Сн(Х) = m(Х)/(ƒэкв(Х).M(Х).V).
Молярная концентрация равна количеству вещества (в молях), содержащегося в 1л раствора. Она обозначается С(Х), иногда (СM) и рассчитывается как отношение количества растворенного вещества Х к объему V раствора в литрах: С(Х)=n(Х)/V=m(Х)/M(Х).V. При записи молярной концентрации используют, например. следующие формы: раствор с молярной концентрацией НСl, равной 0,1моль/л или С(НСl) =0,1 моль/л, 0,1 М раствор НСl (децимолярный раствор НСl) или 0,1 М НСl (здесь буквой М обозначают «молярный»). Все эти записи означают, что в 1л раствора содержится 0,1моль НСl.
При записи молярной концентрации эквивалента, например, для КMnО4 в полуреакции MnО4-+8Н++5е-=Mn2+ +4 Н2О, используют такие формы: С(1/5 КMnО4) = 0,1моль/л, 0,1 н. или 0,1N раствор КMnО4. Если численные значения молярной концентрации и нормальности совпадают (это наблюдается в тех случаях, когда ƒэкв(Х)=1), то употребляют слово «молярный». Например, для 1М раствора КОН не следует применять выражение 1 н. КОН, а нужно использовать выражение 1 М КОН. Количественная связь между молярной концентрацией вещества и его молярной концентрацией эквивалента выражается уравнением С(Х)/Сн(Х)=ƒэкв(Х). Количество вещества (Х), а следовательно, и его масса в объеме V (л) раствора могут быть рассчитаны как на основе молярной концентрации раствора, так и на основе его нормальности.
В тех случаях, когда речь идет об отношении массы (или объема, или количества вещества) всей системы, термин «концентрация» не употребляют, а говорят о «доле» – массовой, объемной или молярной, и выражают эту долю либо дробью, либо в процентах, принимая систему за 1 или за 100%. Все виды долей в отличие от видов концентрации представляют собой безразмерные величины.
Для обозначения доли компонента приняты следующие греческие буквы: массовая доля ω (омега), объемная доля φ (фи), мольная доля χ (хи), причем: ω((Х)=m(Х)/m, φ(Х)=V(Х)/V, χ(Х) =n(Х)/Σn, где m(Х), m – массы компонента и всей системы соответственно; V(Х), V – объемы компонента и всей системы; n(Х), Σn – количество вещества компонента и всей системы. Следует говорить: «Раствор с массовой долей растворенного вещества 20%». Моляльность раствора – количество вещества (в молях), растворенное в 1 кг растворителя, обозначается Сm(Х) и рассчитывается как отношение количества вещества (Х) к массе растворителя в килограммах: Сm(Х)= n (Х)/m(Υ)=m(Х)/M(Х).mΥ. Титр – ранее использовавшийся способ выражения состава раствора – показывает число граммов растворенного вещества в 1мл раствора.
Раствор точно известной концентрации называется стандартным раствором или титрантом, а процесс постепенного добавления его к анализируемой пробе – титрованием. Момент завершения реакции между титрантом и определяемым веществом называется точкой стехиометричности (точкой эквивалентности).
К реакциям, протекающим в стехиометрических отношениях, применим закон эквивалентов. Поэтому, если реакция проведена до конца, число эквивалентов определяемого компонента равно числу эквивалентов реагента. Т. е. моль эквивалента любой кислоты способен нейтрализовать моль эквивалента любого основания. Таким образом, и при титровании, поскольку его заканчивают в точке эквивалентности, всегда затрачиваются одинаковые количества эквивалента титруемого и титрующего веществ. Например, в точке эквивалентности титрования раствора Н2SО4 раствором щелочи (Н2SО4 +2NаОН =Nа2SО4 +2Н2О) количества эквивалентов Н2SО4 и NаОН равны между собой: n(1/2Н2SО4=n(NаОН).
Методы титриметрического анализа.
В точке эквивалентности количество вещества щелочи, израсходованное на реакцию, всегда точно равно количеству вещества кислоты в анализируемом растворе, например: n(NаОН)=n(НСl) или в общем виде, для любых реагирующих веществ по закону эквивалентов: n1=n2. Для понимания сущности титриметрического анализа и связанных с ним расчетов важное значение имеет уравнение: с(NаОН)V(NаОН)=с(НС1)V(НС1), где с –нормальные концентрации растворов; V – их объемы. Общее уравнение имеет вид с1V1=с2 V2. Если известен объем одного из растворов, то из этого уравнения можно вычислить его концентрацию: с1=с2V2/V1 или с2=с1V1/ V2.Точку эквивалентности обычно устанавливают по изменению окраски индикатора (индикаторный способ), но иногда прибегают к измерению электрической проводимости или других свойств раствора (физико-химические) способы. Достигнув точки эквивалентности, титрование прекращают. По затраченному объему титранта и его концентрации вычисляют результаты анализа. Предположим, необходимо определить содержание гидроксида натрия в растворе. Для этого отмеряем в коническую колбу точный объем анализируемого раствора (например, 10,00 мл) и титруем из бюретки титрантом – титрованным раствором НС1. В примере титр кислоты равен 0,002302г/мл и до достижения точки эквивалентности добавили 17,50 мл раствора кислоты. Следовательно, на реакцию израсходовали 17,50.0)002302=0,04029 г НС1. По уравнению реакции нетрудно вычислить какой массе NаОН, находящегося в растворе, соответствует эта масса кислоты: 40 г – 36,5, а 0,04029 – х, получится 0,03676 г. Преимущество титриметрического анализа перед гравиметрическим состоит в скорости, т. е. быстроте определения. В гравиметрическом анализе осаждение (выполнение реакции) для получения осаждаемой формы является лишь началом определения, в титриметрическом анализе выполнение реакции (титрование) и заканчивает определение. Практически одинакова и точность этих анализов. В титриметрическом анализе применяются химические реакции различных типов и в различных практических областях в агрохимии, в пищевых лабораториях, в фармацевтических и различных производственных лабораториях. По типу используемых химических реакций методы титриметрического анализа разделяют на три группы: 1. методы, основанные реакциях соединения ионов; 2. реакциях окисления-восстановления; 3 на реакциях комплексообразования. К 1-ой группе относят методы кислотно-основного и осадительного титрования, ко второй – различные методы окислительно-восстановительного титрования и к 3-ей – методы комплексометрического (хелатометрического) титрования. Метод кислотно-основного титрования (нейтрализации) основан на взаимодействии кислот с основаниями или на взаимодействии протонов с гидроксидионами: Н+ + ОН- = Н2О +57,32Дж. Метод позволяет определять в растворах не только концентрацию кислот и оснований, но и концентрацию гидролизующихся солей. Для определения в растворах концентрации оснований или солей, дающих при протолизе щелочную реакцию, используют титрованные растворы кислот – ацидиметрию. Концентрацию кислот или гидролитически кислых солей определяют с помощью титрованных растворов сильных оснований – алкалиметрия. Точку эквивалентности при нейтрализации определяют по изменению окраски индикатора. Метод осадительного титрования – определяемый элемент при взаимодействии с титрованным раствором, может осаждаться в виде малорастворимого соединения. Последнее, изменяя свойства среды, позволяет тем или иным способом определить точку эквивалентности – аргентометрия с индикатором хроматом калия для определения ионов хлора, до выпадения кирпичнокрасного осадка хромата серебра. В этот момент титрование прекращают и ведут расчет. Название метода определяется раствором титранта: аргентометрия, тиоцианатометрия NН4SСN.
Стандартные и стандартизированные растворы.
Титрованные растворы могут быть получены разными способами и поэтому различают стандартные (приготовленные) и стандартизированные (установленные) р-ры. Стандартные р-ры. Точную навеску в-ва переносят в мерную колбу определенной емкости, растворяют и доводят объем р-ра водой до метки. В этом случае титр р-ра равен навеске (m, г), деленной на объем р-ра (V, мл): Т = m/V. Чтобы перейти к молярной концентрации эквивалента (с), достаточно титр рас-ра умножить на 1000 и разделить на молярную массу эквивалента растворенного в-ва. Например, если 0,5312 г карбоната натрия Nа2СО3 растворили в мерной колбе вместимостью 100 мл, то титр раствора равен Т=0,5312/100=0,005312 г/мл, а молярная концентрация эквивалента с=0,005312· 1000/53,0=0,1002. Титрованные растворы, полученные по точной навеске вещества, называют стандартными (приготовленными). Подобным способом могут быть приготовлены р-ры, отвечающие определенным требованиям: они должны быть хч; устойчивыми при хранении как в твердом виде, так и в растворе; состав их должен строго соответствовать определенной химической формуле. Такие р-ры называют первичными стандартами для установления титра других рабочих растворов. Примеры, для установки титра кислот служат тетраборат натрия Nа2В4О7· 10Н2О и карбонат натрия Nа2СО3. Титры (или нормальные концентрации) растворов оснований устанавливают по таким исходным стандартным веществам, как щавелевая Н2С2О42Н2О или янтарная Н2С4Н4О4. Таким образом, теперь следует объяснить, что такое стандартизированные растворы. Как можнопонять из вышесказанного к ним относятся вещества, не обладающие вышеперечисленными свойствами стандартных растворов. Это минеральные кислоты, щелочи, перманганат калия, тиосульфат натрия. Точные их растворы не возможно приготовить по навеске, т. к. продажные минеральные к-ты имеют непостоянный состав, а выделить их в чистом виде невозможно. А щелочи NаОН и КОН даже при взвешивании поглощают С (1V) и воду из воздуха, т. е. изменяют свой состав Перманганат калия и тиосульфат изменяются при растворении, взаимодействуя с примесями воды. Следовательно, содержание их в растворе не может соответствовать навеске. Поэтому аналитики готовят растворы этих веществ, взвешивая приблизительно необходимое количество реагента и растворяя навеску в необходимом объеме воды. Затем титруют подходящим раствором стандартного вещества. Зная объем, пошедший на титрование, и концентрацию стандартного вещества – известную концентрацию раствора первичного стандарта, вычисляют нормальную концентрацию и титр стандар тизируемого. Растворы, титр которых находят не по точной навеске, а устанавливают по стандартному веществу, называют установленными или стандартизированными (растворами вторичных стандартов). Для соляной к-ты по раствору тетрабората натрия, для гидроксида натрия по стандартному раствору щавелевой к-ты. Применяют идругие приемы, например, устанавливают титр р-ра НС1 путем титрования раствором натрийя гидроксида, если нормальная его концентрация установлена предварительно по соответствующему стандартному веществу. Раствор соляной кислоты после такого определения титра может быть также использован для дальнейшего установления титра других веществ. Этот прием очень удобен в практике, т.к. не надо каждый раз готовить стандартные разные растворы, но недостаточно точен и вносит иногда значительную погрешность в анализ. Следовательно, титрованные стандартные растворы следует готовить очень тщательно и пользоваться одной и той же мерной посудой как при их приготовлении, так и при проведении анализа, т. е. в одинаковых условиях. В практике титрованные растворы готовят из фиксаналов или стандарт-титров. Это запаянная стеклянная ампула содержащая количество вещества необходимое для приготовления 1 л точно 0,1 н или 0,01н раствора. Фиксаналы выпускаются в виде жидких (минеральные к-ты, едкие щелочи) и сухих (перманганат калия, карбонат или оксалат натрия и др.). Рассказать как готовят раствор из фиксанала (боек, воронка, промывалка). Титрованные растворы обычно хранятся несколько месяцев, если нет специальных указаний, особенно требовательны в этом отношении едкие щелочи, при хранении их необходимо предохранять от контакта с воздухом, их хранят в полиэтиленовой плотно закрытой посуде. Для титрования стандартными растворами есть специальные столы и титровальные установки, которые используются при серийных анализах. При этом стандартные или установочные растворы не контактируют с воздухом и с помощью специальных сифонов раствор поступает в бюретку, есть специальные автоматические бюретки. При пользовании мерной посудой существуют определенные правила пользования и мойки. Для бюреток: каждое титрование начинают с конечного деления шкалы, т. к. при этом лучше всего компенсируются погрешности калибрования бюретки. Выпускают раствор из бюретки не очень быстро (не более 3-4 капель в секунду). Иначе раствор не будет вовремя стекать со стенок и отсчет окажется неверным. Объем расходуемого на тирование р-ра не должен превышать вместимость одной бюретки и не превышать 20 мл для 25 мл бюретки и 30 мл для бюретки на 50 мл. Ошибка на +- 0,02 мл составит 0,02·100 / 10мл = 0,2%. После окончания работы сливают раствор из бюретки, моют, наполняют дистиллированной водой и накрывают пробиркой для предохранения от пыли. Правила заполнения раствором бюретки и удаление пузырьков воздуха. Пипетки. Не следует выдувать или вытряхивать последние капли жидкости из пипетки, моют пипетки обычным способом, перед употреблением ополаскивают рабочим раствором, с которым будут работать. Проверка мерной посуды взвешиванием и бюреток по 5 мл р-ра и с учетом плотности воды и поправки на температуру. Вычисления в титриметрическом а нализе. Главный принцип: вещества реагируют друг с другом всегда в эквивалентных количествах. Так на титрование до точки эквивалентности всегда расходуется одинаковое число эквивалентных масс кислоты и основания. Следовательно при одинаковой нормальной концентрации растворов реагирующих веществ реакции идут между их равными объемами. Например, на титрование 10 мл 0,1н. р-ра всякой кислоты расходуется такой же объем 0,1 н. р-ра любой щелочи. В этом также удобство использования нормальных растворов. Результаты титриметрического анализа вычисляют тремя способами: 1. Вычисление при выражении концентрации раствора через титр по определяемому веществу; 2. При титровании по методу пипетирования; 3. При титровании методом отдельных навесок. По первому методу, например, Т(АgNО3/Сl-) показывает, сколько граммов С1- осаждается 1 мл р-ра нитрата серебра. В примере концентрация р-ра нитрата серебра равна 0,1100. Тогда 1 мл его содержит 0,1100: 1000 молярных масс эквивалентов нитрата серебра и взаимодействует с таким же числом молярных масс эквивалента иона хлора. Молярная масса эквивалента хлора равна 35,46 г/моль, вычисляем титр нитрата серебра по хлору: Т(АgNО3/Сl-) = 0,1100. 35,46/1000=0,003901 г/моль. На титрование израсходовали 15,00 мл р-ра нитрата серебра, то титруемый р-р содержит хлора: m (С1-)=Т(АgNО3/Сl-)V(АgNО3) = 0,003901·15,00 = 0,05851 г. При вычислениях по нормальности растворов ход вычислений зависит от метода титрования. Метод пипетирования состоит в том, что навеску анализируемого вещества растворяют в мерной колбе, доводят объем водой до метки и для титрования берут определенные (аликвотные) порции р-ра пипеткой. Для расчета используют уравнение с1V1=с2V2. Задача: в мерную колбу внесли 0,6504 г продажной щавелевой к-ты. Растворили и довели объем до метки 100 мл. Пипеткой брали по 10 мл полученного р-ра и титровали 0,1026 н. р-ром гидроксида натрия, расход которого составил в среднем 9,85 мл. Определить массовую долю (%) щавелевой к-ты в продажном препарате. Вычисляем нормальную концентрацию раствора щавелевой к-ты: с(Н2С2О42Н2О) 10,00= 0,1026. 9,85; с =0,1026. 9,85/10,00 =0,1011. Затем находим массу щавелевой к-ты в 0,1 л р-ра. При этом исходить следует из того, что щавелевая к-та превращается в щавелевокислый натрий, следовательно, молярная масса эквивалента ее равна ½ молярной массы, т.е.126,6: 2=63,03 г/моль. Поэтому m=0,1011. 63,03.0,1=0,6372 г. Следовательно, массовая доля (%) щавелевой к-ты равна: ω (Н2С2О42Н2О)= m (Н2С2О42Н2О)/ m(навески)= 0,6372/0,6504=0,9797, или 97,97%. В случае использования метода отдельных навесок уравнение с1V1=с2V2 не может быть использовано, т. к. навеску растворяют в произвольном объеме воды. Вычисляют результат, исходя из того, что при титровании в-ва взаимодействуют эквивалентными количествами. Какова массовая доля (%) Н2С2О42Н2О в образце щавелевой к-ты, если на титрование 0,1500 г его пошло 25,60 мл 0,09002 н. р-ра NаОН? Вычисляют, сколько молярных масс эквивалента NаОН содержалось в 25,60 мл раствора и участвовало в реакции: 1000 мл р-ра NаОН содержит 0,09002 молярных масс эквивалента NаОН, а 25,60 мл NаОН содержат х молярных масс NаОН, Х = 25,60. 0,09002 / 1000 = 0,002305 молярных масс эквивалента NаОН. Но при титровании 1 эквивалент в-ва взаимодействует с 1 эквивалентом другого. Т. е. Это означает, что в реакции участвовало также 0,002305 молярных масс эквивалента Н2С2О42Н2О. Молярная масса эквивалента щавелевой к-ты равна 63,03 г/моль. Следовательно, анализируемый раствор содержит 0,002305. 63,03 = 0,1453 г Н2С2О42Н2О. В массовых долях (%) это составляет: ω (Н2С2О42Н2О) = m (Н2С2О42Н2О)/m (образца) = 1,1435 г / 0,1500 г = 0,9686 (96,86%). Во многих случаях для уточнения результатов анализа (из-за неточности навесок при приготовлении растворов или исследуемого вещества) вводят понятие поправочного коэффициента. Поправочный коэффициент К равен отношению практически взятой навески к теоретически вычисленной К = С/ С1, где С – нормальность приготовленного раствора; С1 – теоретическая нормальность. Например, требуется приготовить р-р с заданной (теоретической) концентрацией 0,1000 моль/л. Практически, приготовлен раствор с близкой концентрацией 0, 1056 моль/л. Тогда находим поправочный коэффициент К = 0,1056 / 0,1000 = 1,056. Поэтому практически во всех титриметрических анализах, в расчетах вводится поправочный коэффициент для данного раствора. При массовых анализах и использовании нового раствора (вновь приготовленного) вводится новый поправочный коэффициент, при правильном приготовлении растворов он должен быть в пределах 0,98 – 1,02. Следовательно, для расчетов следующие основные правила: 1. Титры р-ров, выраженные в граммах различных в-в относятся друг к другу как молярные массы эквивалентов этих в-в: ТН2SО4/ NаОН/ТН2SО4=Э NаОН/ЭН 2 SО 4, ТН 2 SО 4 / NаОН = ТН2SО4.ЭNаОН/ЭН2 SО4, где Т Н2 SО4/NаОН – титр по определяемому в-ву, т. е. количество граммов определяемого в-ва, оттитрованное 1 мл определяемого раствора. При одинаковой концентрации р-ов реакции идут между равными объемами их: V1/ V2=С2/С1 или V1/ V2= N2/ N1. Массу навески в-ва можно вычислить по формуле: q = СVЭ / 1000.
ТЕМА 9
МЕТОДЫ КИСЛОТНО-ОСНОВНОГО ТИТРОВАНИЯ
Кислотно – основное титрование, реакции нейтрализации, скачок титрования, построение кривых титрования, правила выбора индикатора при кислотно – основном титровании, требования, предъявляемые к индикаторам, методы амперометрического, кондуктометрического и потенциометрического титрования. Кривая титрования – графическое изображение зависимости изменения концентрации С (Х), определяемого в-ва Х или некоторого связанного с ним свойства системы (раствора) от объема V(Т) прибавленного титранта (Т). По оси абсцисс откладывают объем прибавляемого титранта, по оси ординат соответствующее значение рН. По тенциометрическое тирование – изменение концентрации иона непременно сопровождается изменением потенциала на индикаторном электроде, погруженном в титруемый р-р. При этом около точки эквивалентности наблюдается скачок потенциала, который фиксируется при помощи потенциометра. Кондуктометрическое титрование – изменение электрической проводимости титруемой среды между 2-мя инертными электродами. Амперометрическое титрование – за ходом титрования следят с помощью ртутного капающего, вращающегося платинового или другого микроэлектрода, играющего роль индикаторного электрода и находящегося в паре с подходящим электродом сравнения. Кулонометрическое титрование -измерение количества электричества, затраченного на выполнение электродной реакции. При этом титрующий агент (например. кислотно-основного типа) не прибавляется в виде стандартного р-ра, а образуется в р-те электродной реакции, т. е в результате электролиза при постоянном токе. Титрование сильной к-ты сильным основанием: НС1+КОН =КС1+Н2О, в процессе титрования происходит постоянная, непрерывная нейтрализация к-ты. В точке «100» наступает полная нейтрализация. Т. к. соль сильного основания и сильной к-ты не способна к гидролизу, рН в этой точке равен 7 (кривая титрования на обороте). При дальнейшем титровании в р-ре накапливается избыток щелочи и в этом случае рН р-ра рассчитывается по формулам 1 и 2. Титрование слабой к-ты сильным основанием: СН3СООН+КОН=СН3СООК+Н2О, в нулевой точке рН вычисляют по формуле 3. По мере титрования в среде накапливается ацетат натрия. Смесь слабой к-ты и ее соли представляет собой кислотный буферный р-р. Поэтому в интервале 10-99 мл прибавленного титранта рН рассчитывают по формуле 5. В точке 100 вся к-та полностью связана в соль. Соль слабой к-ты и сильного основания легко гидролизуется с отщеплением свободных гидроксилов. Поэтому точка эквивалентности находится в щелочной области (кривая титрования на обороте); рН рассчитывается по формуле 8. При дальнейшем титровании в растворе накапливается избыток титранта и рН в области 101-200 мл прибавленного титранта рассчитывают по формуле. Титрование слабого основания сильной к-той: NН4ОН+НС1=NН4С1+Н2О, в нулевой точке рН раствора вычисляют по формуле 4. По мере титрования в р-ре накапливается соль – хлорид аммония. Смесь слабого основания и его соли представляет собой щелочной буферный р-р, поэтому расчет рН в интервале 10-99 мл прибавленного титранта проводят по формуле 6. В точке эквивалентности (точка 100) все основание переходит в соль. Соль слабого основания и сильной к-ты легко гидролизуется с отщеплением свободных протонов. Поэтому точка эквивалентности находится в кислой области (кривая тирования на обороте), расчет рН ведут по формуле 7. При дальнейшем титровании в р-ре накапливается избыток титранта и рН в интервале 101-200 рассчитывается по формуле 1. Титрование слабой к-ты слабым основанием: СН3СООН+NН4ОН =СН3СООNН4+Н2О. В нулевой точке рН раствора находят по формуле 3. В интервале 10-99 мл прибавленного титранта смесь представляет собой кислотный буфер и рН р-ра рассчитывают по формуле 5. В точке эквивалентности вся кислота связана в соль. Соль слабой к-ты и слабого основания полностью гидролизуется с образованием слабых к-ты и основания. В зависимости от значений рКα и рКb рН р-ра соли может находиться как в кислой, так и в щелочной области (кривая титрования); рН в этой точке рассчитывается по формуле 9. При дальнейшем тировании рН в точке 101 находят по формуле 4. После того как в растворе накопится достаточный избыток титранта, титруемая смесь будет представлять собой щелочной буфер (смесь слабого основания и его соли) и рН в интервале 110-200 мл вычисляют по формуле 6. Как мы уже говорили ацидиметрия – ацидиметрическое тирование – это метод определения сильных и слабых оснований, солей слабых к-т, основных солей и других соединений, обладающих основными свойствами, путем титрования стандартным р-ром сильной к-ты. При титровании сильных оснований протекает реакция: ОН-+Н3О+=2Н2О, среда в ТЭ – нейтральная. При титровании слабых оснований, например аммиака, NН3+Н3О+= NН4++Н2О образуются катионы слабого основания, подвергающиеся гидролизу: NН4++Н2О= NН3+Н3О+ поэтому среда в ТЭ – слабокислая, рН<7. При титровании солей слабых одноосновных к-т, например ацетатов, СН3СОО-+Н3О+= СН3СООН+Н2О в ТЭ в р-ре присутствует слабая к-та, вследствие диссоциации которой р-р имеет слабокислую реакцию, рН<7. При титровании солей слабых 2-хосновных к-т, например карбонатов, после присоединения к аниону к-ты одного протона образуется кислый анион слабой к-ты СО32-+Н3О+=НСО3-+Н2О, подвергающийся гидролизу: НСО3-+Н2О=Н2СО3+ОН-, вследствие чего реакция среды в 1-ой ТЭ –слабощелочная, рН>7. При продолжении титрования кислого аниона слабой 2-хосновной к-ты во 2-ой ТЭ присутствует эта слабая к-та: НСО3-+Н3О+=Н2СО3 вследствие частичной диссоциации которой среда во 2-ой ТЭ – слабокислая, рН<7. Т.о. при ацидиметрическом титровании среда в ТЭ может быть нейтральной, слабо щелочной или слабокислой в зависимости от природы титруемого вещества. Алкалиметрия (алкалиметрическое титрование) – метод определения сильных и слабых к-т, кислых солей, солей слабых оснований путем титрования стандартным раствором сильного основания. При титровании сильных к-т реакция среды в ТЭ – нейтральная. При титровании слабых 1-ноосновных реакция среды в ТЭ слабощелочная вследствие гидролиза образующихся анионов слабой кислоты, например: СН3СООН+ОН-=СН3СОО-+Н2О(титрование), СН3СОО-+Н2О=СН3СООН+ОН-(гидролиз). При титровании многоосновных к-т реакция среды в разных ТЭ неодинакова и зависит от констант кислотной диссоциации последовательно образующихся в результате гидролиза к-т (к-ты средней силы дают кислую реакцию среды), например титрование Н2ЅО3+ОН-=НЅО3-+Н2О; амфолит НЅО3- проявляет как кислотные, так и основные свойства. Пос
|
|
|
|
|
Дата добавления: 2013-12-12; Просмотров: 2382; Нарушение авторских прав?; Мы поможем в написании вашей работы!