КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Блиц –· тест
|
|
|
|
|
|
|
|
|
|
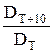 = 2 + 4
= 2 + 4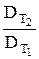 = γ
= γ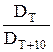 = γ
= γ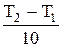
|
|
- Что называется коэффициентом массопередачи? Коэффициентом массопередачи называется:
- скорость реагентов при градиенте концентрации равной единице
- скорость переноса массы реагентов при изменении концентрации на единицу
- перенос массы реагентов в единицу времени
- скорость переноса массы реагентов в ходе гетерогенного процесса скорость подвода и отвода реагентов в реакционную зону
Литература:
- Теория металлургических процессов: учебник для вузов. Рыжонков Д.И., Арсентьев П.П., Яковлев В.В. и др. - М.: Металлургия, 1989, 392 с.
- Теория металлургических процессов: учебное пособие для вузов. Попель С.И., Сотников А.И., Бороненков В.И. М.: Металлургия, 1986. с.483
- Теория металлургических процессов. С.И. Филиппов.
М: Металлургия, 1967 – 279 с
|
|
|
Борнацский И.И. Теория металлургических процессов, учебное пособие. Киев, Донецк Высш.шк. 1978, 288 с.
- Казачков Е.А. Расчеты по теории металлургических процессов, Ю М., «Металлургия», 1986, 288 с.
- Симбинова К.Ж., Байсанов С.О., Никитин Г.Н. Физико-химия металлургических систем и процессов. Алматы 1993 г.
- Симбинов Р.Д., Симбинова К.Ж. Исследование вязкости жидкостей и оксидных расплавов. Актобе 2005 год
- Термодинамика и кинетика процессов диссоциации карбонатов и оксидов: лабораторный практикум. Симбинов Р.Д., Симбинова К.Ж. Актобе 2005 год.
- Симбинова К.Ж. Методические рекомендации по выполнению лабораторных работ. Алма-Ата, 1990, 75 с.
Лекция №7 «Взаимодействие сульфидов с газами, металлами и оксидами»
План лекции:
1.Взаимодействие твердых и газообразных фаз в системе Ме-S-О. Образование и диссоциация сульфатов.
2.Термодинамические характеристики процесса.
3.Взаимодействие в системах Ме-Ме-S.
4.Высокотемпературные процессы взаимодействия в системе Ме-S-О.
5.Термодинамические условия. диаграммы парциальных давлений в системах Сu-S-О, РЬ- S-О. Взаимодействие в системе Mе-Ме-S-О.
6.Механизм и кинетика взаимодействия в системе Ме-S-О при наличии твердых фаз. 7.Особенности механизма и кинетики взаимодействия в системе Ме-S-О при наличии жидких фаз.
Цель лекции
Ознакомление с основными взаимодействия сульфидов с газами, с металлами и оксидами
Дидактические единицы:
Сульфиды, шлак; вязкость; кислотность; основность шлака
1. Процессы диссоциации и образования карбонатов, окислов и других химических соединений сопровождаются фазовыми превращениями. Всякое фазовое превращение происходит в результате возникновения в старой (исходной фазе) небольших объемов новой фазы, называемых зародышевыми центрами, и последующего их роста'. Чем больше возникает зародышевых центров и болыпе скорость их роста, тем быстрее развивается фазовое превращение.
|
|
|
При возникновении участков новой фазы свободная энергия системы изменяется под влиянием двух взаимопротивоположных процессов — с одной стороны, часть молекул, атомов, ионов из старой фазы, обладающих более высоким уровнем свободной энергии, переходит в новую фазу с меньшим уровнем свободной энергии. С другой стороны, благодаря возникновению межфазной поверхности раздела свободная энергия системы увеличивается.
Отклонения тех или иных параметров системы от их наиболлее вероятных значений в состоянии энергитического (теплового) равновесия в различных точках системы называается флуктуацией.
Различают гомофазные и гетерофазные флуктуации.
Когда отклонения плотности или концентрации в системе не приводят к изменению фазового состояния, то такие флуктуации называются гомофазными.
Если в небольших объемах системы происходят отклонения, вызывающие возникновение частиц новой фазы, то такие флуктуации называется гетерофазными.
Возможность гомофазных и гетерофазных флуктуаций Р определяется следующим выражением:
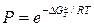 (1)
(1)
где А — коэффициент пропорциональности; 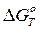 — изменение изобарного потенциала системы, обусловленное появлением флуктуации.
— изменение изобарного потенциала системы, обусловленное появлением флуктуации.
Для процессов диссоциации карбонатов, окислов и сульфидов, при которых образуются новые твердые фазы, характерно возникновение гетерофазных флуктуаций. В основу термодинамического анализа образования зародышей МеО при диссоциации МеСО3 положены результаты работы Я. И. Френкеля. Начальная стадия разложения карбоната протекает без возникновения новой фазы и ограничивается образованием раствора МеО в МеСО3. В зависимости от концентрации растворяющегося вещества растворы разделяются на ненасыщенные, насыщенные и перенасыщенные.
Выделение МеО в самостоятельную фазу возможно только из перенасыщенного раствора. Для термодинамического анализа возникновения зародыша новой фазы МеО приняты следующие обозначения:
l— ребро зародыша новой фазы МеО, имеющего форму кубика;
μо — химический потенциал МеО в новой фазе или в насыщенном растворе МеО в МеС03;
μ — химический потенциал раствора МеО в МеСО3;
|
|
|
σс-н—поверхностное натяжение на границе фаз старая
— новая фаза;
п —число элементарных частиц СаО в единице объема
новой фазы.
Появление зародыша МеО сопровождается возникновением поверхности F = 6 l 2 и соответствующим увеличением термодинамического потенциала на Fσс-н- В целом изменение термодинамического потенциала системы описывается уравнением
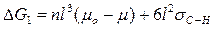 (2)
(2)
где ΔG1 — величина термодинамического потенциала, рассчитана на один зародыш.
Первый член уравнения учитывает изменение изобарного потенциала при образовании зародыша новой фазы. Величина и знак разности μo—μ при данной температуре определяются концентрацией раствора. Для ненасыщенного раствора μ—μo, а разность μo—μ >0, т. е. первое слагаемое в уравнении (2) •— положительная величина. Второе слагаемое — всегда положительная величина. Таким образом, с увеличением размера зародыша функция ΔG1 сохраняет положительные значения (кривая 1 рис. 1).
Повышение концентрации МеО в растворе (карбонате) сопровождается увеличением химического потенциала μ и приближением его значений к μo: при этом разность μo—μ убывает. Качественных изменений еще не происходит, уменьшается только темп роста с увеличением l (кривая 2 ).
С течением времени раствор МеО в МеСО3 приближается к насыщению, возрастает вероятность появления флуктуаций, увеличивается число и размер появляющихся и исчезающих зародышей. Вместе с тем пока разность μo—μ, не меняет знака, отсутствуют условия для выделения МеО в самостоятельную фазу.
Если μ—μo, первое слагаемое в уравнении (2) становится отрицательным и при определенной величине l на кривой 3 изменения ΔG появляется экстремум.
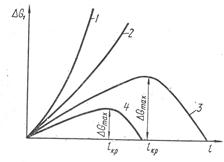
Рис. 1. Изменение термодинами ческого потенциала раствора при образовании зародыша новой фазы:
Такое значение l называется критическим. Для нахождения соответствующего максимума l кр необходимо продифференцировать уравнение (2) и приравнять первую производную нулю:
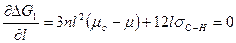 (3)
(3)
Из уравнения (3) получим
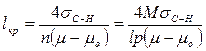 (4)
(4)
где М — молекулярная масса частиц, образующих зародыш; ρ — плотность зародыша; п = ρ/М — число молекул в зародыше.
|
|
|
Величина ΔG1тахопределяет работу, затраченную на образование зародыша критического размера. Она может быть получена после подстановки значений lкр в уравнение (3):
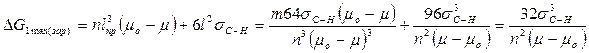
Из уравнения (5) следует, что работа образования зародыша составляет одну треть работы образования поверхности зародыша критического размера:
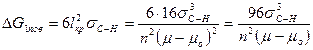
Таким образом,
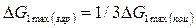 (6)
(6)
Из уравнения (4) следует, что при небольших перенасыщениях, когда разность μ—μo, не намного отличается от нуля, зародыш критического размера имеет значительную величину. С увеличением степени переохлаждения размер критического зародыша уменыпается.
2. Скорость разложения веществ изменяется с течением времени τ по сложной зависимости. На рис. 2 показаны два возможных вида кинетических кривых диссоциации химических соединений. Первый вид характеризует нарастание скорости реакции во времени с последующим прохождением через максимум т и S-образный ход кривой степени превращения (кривая 1). Второй вид дает представление о таких прoцессах, когда скорость разложения снижается по ходу диссоциации (кривая 2). Для первого вида кинетических кривых процесс диссоциации делится на три периода. Первый период называется инкубационным (область I), второй — автокаталитичесkим (область II) и третий — периодом усредненного фронта (область III). В инкубационном периоде в отдельных активных центрах зарождаются кристаллики новой фазы и возникает поверхность раздела между старой и новой фазами.
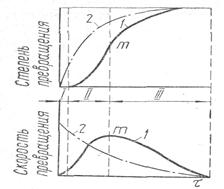
Рис 2. Возможные виды кинетических кривых диссоциации химических соединений.
После появления зародышей новой фазы развитие процесса во II области заметно облегчается. Зародыши кристаллов новой фазы увеличиваются в результате последовательного покрытия граней мономолекулярными слоями продуктов диссоциации. Скорость процесса возрастает вместе с суммарной поверхностью раздела. После того, как скорость превращения достигнет максимума, наступает период усредненного фронта.
Зародыши новой фазы возникают с большей вероятностью тогда, когда значения энергии активации процесса для преодоления энергетического барьера небольшие. Кинетика роста числа зародышей и образование продуктов диссоциации твердых веществ описываются общим уравнением
V= Ce-ΔGкр/kTe-E/kT (1)
где ΔGкр — значения изобарного потенциала для зародыша новой фазы критического размера; Е — величина энергии активации, обеспечивающая перемещение частиц к месту формирования новой фазы; k — константа Больцмана.
В уравнении (1) первый экспоненциальный множитель 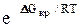 характеризует скорость образования зародыша, второй — скорость роста зародышей. В целом уравнение (1) дает представление об особенностях топохимических реакций, в том числе реакций диссоциации карбонатов и окислов.
характеризует скорость образования зародыша, второй — скорость роста зародышей. В целом уравнение (1) дает представление об особенностях топохимических реакций, в том числе реакций диссоциации карбонатов и окислов.
При оценке режима гетерогенных процессов можно пользоваться полуэмпирическими кинетическими уравнениями типа
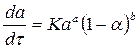 (2)
(2)
где α — степень превращения вещества; К —константа скорости; a и в — постоянные величины. Обычно величина а не превышает единицы, а в может быть как больше, так и меньше единицы. Если а ≤1 и в= 1, то уравнение (2) можно рассматривать как приближенную дифференциальную форму выражения, полученного Б. В. Ерофеевым
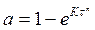 (3)
(3)
где п — связано с а отношением (п — 1)/п = а.
Применимость уравнения (3) для оценки режима и механизма гетерогенных процессов исследовали С. А. Казеев, А. Н. Колмогоров, Б. В. Ерофеев. Значения п могут быть больше или меньше единицы в зависимости от характера процесса. Принято считать, что при п≥1 процесс осуществляется в кинетическом режиме. Переход процесса в диффузионную область характеризуется уменьшением значения п. Тогда п≈0,5, процесс протекает в диффузионном режиме.
После двойного логарифмирования уравнения (3) получим
где
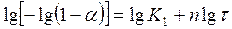
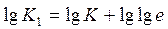 (4)
(4)
При помощи выражения (4) по экспериментальным данным можно определять значение п. Для этого на оси абсцисс откладываются значения l gτ и на оси ординат — lg[—lg(1—α)]. При соответствии процесса уравнению (3) получается прямая линия, наклон которой определяет значения п.
Для оценки режима гетерогенного процесса, кроме уравнения (3), используются другие соотношения, представляющие частные случаи указанного кинетического уравнения. K их числу относятся соотношения
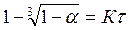 (5)
(5)
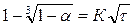 (6)
(6)
первое из которых характерно для процессов, протекающих в кинетической области, а второе — в диффузионной.
§3. Процесс диссоциации карбонатов складывается из трех звеньев: кристаллохимического превращения, внутренней и внешней диффузии. В самом общем виде связь макроскопически наблюдаемой скорости с особенностями процессов в отдельных звеньях описывается уравнением
 (1)
(1)
где Vртах, VDmax и VDmax — соответственно скорости образования СО2, внутреннего и внешнего диффузионного потоков.
Превращения карбонатов в окислы связаны с уменьшением объема вещества. Диссоциация карбонатов возможна, если обеспечивается непрерывный отвод СО2 из реакционной зоны и существуют следующие соотношения:
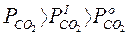
где Рсо2 — упругость диссоциации карбоната; Р 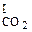 — парциальное давление СО2 в реакционной зоне;
— парциальное давление СО2 в реакционной зоне; 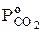 -парциальное давление СО2 в окружающем пространстве.
-парциальное давление СО2 в окружающем пространстве.
Развитие процесса диссоциации связано с продвижением фронта реакции от наружной поверхности карбоната к центральной части и с увеличением слоя образующегося окисла, обладающего значительной пористостью и относительно хорошей газопроницаемостью. В начале процесса диссоциации карбоната, когда толщина окисла металла небольшая, диффузия СО2 через такой слой не сдерживает кристаллохимическое превращение. С течением времени слой образующегося окисла становится значительным и диссоциация карбоната тормозится внутренней диффузией СО2. При определенных условиях диффузия СО2 приобретает значения ведущего звена, что связано с переходом процесса из кинетической в диффузионную область.
Kогда в куске карбоната произошла частичная диссоциация, на его поверхности появляется слой окисла металла толщиной х. В том случае, когда внешнее диффузионное сопротивление близко к нулю (→ - 0), кинетика процесса описывается двумя звеньями —кристаллохимическим превращением и внутренней диффузией. Скорость химической реакции определяется уравнением
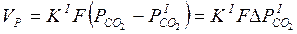 (2)
(2)
где К ' —константа скорости прямой реакции диссоциации карбоната; F—поверхность реакционной зоны; ΔР'со2— разность между упругостью диссоциации и давлением СО2 в реакционной зоне.
Скорость внутренней диффузии можно приближенно рассчитать, пользуясь уравнением
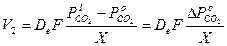 (3)
(3)
где D е — эффективный коэффициент диффузии углекислого газа через пористый слой окисла.
Установившийся процесс диссоциации карбоната характеризуется равенством величин V р и VD:
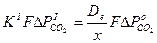
или
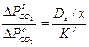 (4)
(4)
После преобразования уравнения (10) получим
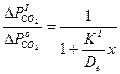 (5)
(5)
Величину ΔР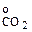 = Δ
= Δ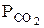 + ΔРС. Т. Ростовцев рассматривает как суммарный перепад давлений СО2, обеспечивающий осуществление процесса в обоих звеньях. Распределение этого перепада между кристаллохимическим и внутридиффузионным звеньями происходит в соответствии с их скоростными возможностями, определяемыми константами К' и De,. Kогда К' «De и отношение ΔР'со2: ΔРблизко к единице, в реакционной зоне устанавливается давление СО2, близкое к тому, которое наблюдается у наружной поверхности куска карбоната.
+ ΔРС. Т. Ростовцев рассматривает как суммарный перепад давлений СО2, обеспечивающий осуществление процесса в обоих звеньях. Распределение этого перепада между кристаллохимическим и внутридиффузионным звеньями происходит в соответствии с их скоростными возможностями, определяемыми константами К' и De,. Kогда К' «De и отношение ΔР'со2: ΔРблизко к единице, в реакционной зоне устанавливается давление СО2, близкое к тому, которое наблюдается у наружной поверхности куска карбоната.
Kогда К' «De при сравнительно небольшой толщине слоя окисла х отношение ΔР'со2: ΔРхарактеризуется небольшими значениями и почти весь суммарный перепад ΔРиспользуется в медленном диффузионном звене, процесс находится в диффузионной области и определяется законом диффузии.
4. С. Т. Ростовцев путем аналитических исследований получил уравнения для определения скорости реакции и времени, необходимого для достижения определенной степени разложения в кусках сферической формы при условии, что теплообмен не лимитирует процесса. Применительно к условиям диссоциации карбонатов этими уравнениями учитываются кристаллохимическое превращение и внутренняя диффузия С02 в карбонате в пределах реакционной зоны и в покровном слое МеО и внешняя диффузия от наружной поверхности кусков карбоната в газовый поток.
Для определения типа процесса можно пользоваться величиной безра-змерного критерия:
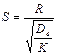 (6)
(6)
или
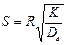 (7)
(7)
где τ — радиус исходного куска карбоната.
Для пористых известняков при низких температурах
K<De, реакционная зона размытая и может распространиться на весь объем куска, процесс протекает в кинетической области. Диссоциация карбонатов с плотной структурой обычно осуществляется при более высоких температурах. Этим условиям отвечает соотношение К»Dе, величина критерия S становится значительной, глубина реакционной зоны сокращается и процесс переходит в диффузионную область.
Кристаллические решетки карбонатов и окислов состоят из ионов. У карбонатов СаС03, МgСО3, FеСО3 в узлах кристаллических решеток расположены катионы Са2+, Мg2+, Fе2+, Мп2+ и анионы СО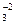 ~, у окислов — соответствующие катионы металлов и анионы кислорода
~, у окислов — соответствующие катионы металлов и анионы кислорода
O-2.
Диссоциация карбонатов начинается с распада тех анионов СО~, в которых молекулы СО2 получили при нагреве дополнительный запас кинетической энергии, необходимый для отрыва от анионов O-2~:
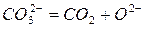
Удаление молекул СО2, обладающих сравнительно большими размерами, из углубленных слоев кристаллической решетки путем диффузии затруднительно. Если же молекулы СО2 длительное время не покидают места своего возникновения, возможно образование исходных анионов СО~.
С развитием процесса диссоциации в поверхностном слое карбоната происходит накопление анионов, что вызывает образование перенасыщенного раствора МеО в МеСО3. Этому способствует незначительная растворимость МеО в МеСО3.
При появлении границы раздела МеО—МеСО3 процесс диссоциации значительно ускоряется и протекает в автокаталитическом режиме. Но такое течение процесса будет продолжаться только определенный промежуток времени. Замедление процесса происходит в результате утолщения внешнего слоя продукта реакции.
На рис. 3 показана термограмма диссоциации СаСОз и карбонизации СаО, из которой следует, что при нагревании карбоната и охлаждении окисла в токе СО2 (давление 101,3 кПа) наблюдались две температурные остановки при 910° С (диссоциация карбоната) и при 888° С (карбонизация окисла), причем продолжительность первой остановки существенно больше. Такая разница в продолжительности остановок связана с тем, что количество поглощеного тепла при диссоциации СаСО3 превышает количество СаО.
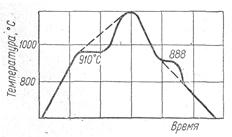
Рис. 3. Термограмма диссоциации СаСОз и карбонизации тепла, выделенного при карбонизации СаО.
Это означает, что в условиях опыта карбоната разлагается большее количество, чем образуется при карбонизации СаСО3. Такие соотношения обусловлены различной структурой покровного слоя твердых продуктов реакции при диссоциации и карбонизации.
При карбонизации СаО продукт реакции СаСО3 занимает больший объем, чем исходный окисел СаО, и на поверхности окиси кальция образуется сплошной плотный покров. Процесс карбонизации протекает с большей скоростью вначале реакции, когда слой продукта реакции еще небольшой. По мере роста толщины поверхностного слоя диффузионное сопротивление проникновению СО2 в глубииные слои куска извести значительно возрастает, что сопровождается снижением скорости карбонизации.
§5. Процессы диссоциации окислов металлов и карбонатов обладают одной и той же физикохимической природой: они относятся к топохимическим реакциям.
В процессах диссоциации окислов можно выделить следующие этапы: химическую реакцию распада окисла с освобождением атомарного кислорода, адсорбированного на границе раздела фаз, молизацию атомов кислорода, десорбцию молекул кислорода, возникновение зародышей новой фазы и их рост.
Экспериментально изучать диссоциацию прочных окислов крайне затруднительно вследствие очень малых значений величин парциальных давлений компонентов паровой фазы. С помощью массспектрометрического эффузионного метода исследован состав паров над кристобалитом в интервале температур 1200-1950 К, при этом обнаружены молекулы двуокиси кремния, моноокиси кремния, молекулярный и атомарный кислород. Результаты измерений парциальных давлений (Па) компонентов пара над кристобалитом приведены ниже:
| T, K | SiO | SiO2 | O2 | O |
| 9,1∙10-4 | 2,1∙10-6 | 6,9∙10-6 | - | |
| 4,5∙10-3 | 1,3∙10-5 | 10,9∙10-6 | 2,9∙10-5 |
Суммарное давление газообразных компонентов над кремнеземом в интервале температур 2000-3000 К определяется из уравнения
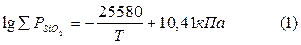
Согласно указанному уравнению давление газообразных компонентов, равное 101,3 кПа, достигается при 3048 К.
С помощью вольфрамовой эффузионной камеры изучались парциальные давления компонентов пара над А12О3(ТВ) при 1992° С. Выше приведены результаты этих исследований:
Т а б л и ц а 3. Значения упругости диссоциации окислов меди СиО и Си2О, Па
| Окисел | Температура, К | ||||
| СиО | 1,2∙10-35 | 2,3∙10-18 | 8,3∙10-10 | 2,4∙10-1 | 3,3∙10-3 |
| Си2О | 4,4∙10-46 | 5,5∙10-27 | 1,8∙10-17 | 4,8∙10-8 | 1,9∙10-4 |
Значительно более высокие величины парциальных давлений продуктов диссоциации наблюдаются у малоустойчивых окислов и перекисей, таких как Аg2О, СаО, Fе2О3, МпО2, ВаО2, СаО2. Из приведенных в таблице данных следует, что при температурах 600 и 900 К упругость диссоциации СиО примерно в 7 раз больше, чем упругость диссоциации Си2О. Равновесная упругости диссоциации СиО была проверена экспериментальным путем. При температурах 900 и 1000° С упругость диссоциации СиО соответственно составляла 1,2-103 и 5,3-103 Па.;Установлено, что скорость диссоциации окиси меди приближенно описывается уравнением
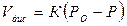 (2)
(2)
где Р0 и Р — равновесное и фактическое давления кислорода в газовой фазе;
К — коэффициент.
Изучение температурной зависимости скорости диссоциации по-казало, что она определяется по уравнению
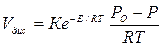 (3)
(3)
Исследована зависимость скорости диссоциации перекисей бария ВаО2 и кальция СаО2 от давления кислорода в газовой фазе. Установлено, что при постоянных температуре и содержании кислорода в твердой фазе скорость диссоциации ВаО2 и СаО2 так же, как и СиО, пропорциональна разности между равновесным давлением 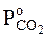 и давлением кислорода в газовой фазе Р. Кажущаяся энергия активации для процесса диссоциации ВаО2 и СаО2 соответственно составляет 192300 и 189000 кДж (кг-моль).
и давлением кислорода в газовой фазе Р. Кажущаяся энергия активации для процесса диссоциации ВаО2 и СаО2 соответственно составляет 192300 и 189000 кДж (кг-моль).
Металлы и сплавы, находящиеся в атмосфере, содержащей кислород или другие кислородсодержащие газы (СО2, Н2О, SО2), покрываются слоем окислов, окалиной, толщина которой увеличивается с течением времени. Этот процесс в области высоких температур относится к высокотемпературной коррозии, протекающей на границе фаз металл — окружающая среда, т. е. является гетерогенным процессом взаимодействия газообразной среды с металлом.
Первопричина коррозии металлов и сплавов — их термодинамическая неустойчивость в различных средах при данных внешних условиях.
Коррозия металлов — это самопроизвольный процесс разрушения металлических материалов вследствие химического или электрохимического взаимодействия их с окружающей средой.
Среди процессов высокотемпературного окисления металлов особое место занимает их окисление кислородом. Этот процесс — самый распространенный вид коррозии металлов в газовых средах при высокой температуре.
Kинетика окисления металлов определяется механизмом их взанмодействия с кислородом. В общем случае суммарный процесс окисления металла включает следующие более простые физические и химические процессы:
— адсорбцию молекул кислорода из газовой фазы на поверхности окисной пленки;
— диссоциацию адсорбированных молекул кислорода на атомы О2→2О;
— ионизацию адсорбированных атомоів кислорода
О + 2е - → О2-
— перемещение (диффузию) ионов кислорода (анионов) в пленке окисла в направлении к металлической поверхности;
— ионизацию атомов металла и переход ионов
(катионов) и электронов
из металлической фазы в пленку окислов
Ме → Меn+ + ne
— перемещение (диффузию) катионов и электронов в пленке окисла в
направлении к границе раздела пленка — газ;
— взаимодействие катионов и анионов с образованием продукта этого взаимодействия — окисла металла
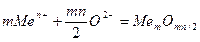
Рассмотрим схему образования окисной пленки на металле (Рис.1). После того как в процессе окисления металлов на поверхности образовалась достаточно толстая пленка окислов, химическая реакция перестает быть контролирующей стадией, эти функции переходят к диффузии тех или иных частиц (катионов, анионов илй электронов).
Зона роста окисной пленки может располагаться на границах газ — пленка, пленка—металл или во внутренних слоях пленки, она определяется скоростью перемещения катионов и анионов. Когда в пленке перемещаются катионы и электроны, зона роста будет находиться на ее внешней стороне (рис. 1, а)
Пленка растет со стороны поверхности металла, если скорость переме-щения анионов существенно выше скорости движения катионов и электронов. При соизме-римых скоростях перемещения анионов, катионов и электронов зона роста располагается внутри пленки.
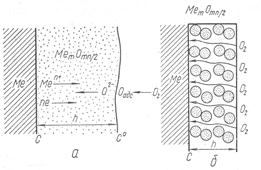
Рис. 1. Схема образования окисной пленки на металле.
Анализ экспериментальных данных показал, что окисление одного и того же металла при разных температурах подчиняется различным кинетическим закономерностям. У большинства металлов наблюдается следующая последовательность смены кинетических закономерностей: логарифмическая, параболическая в промежуточной температурной области, линейная — при высокой температуре.
6. Линейная закономерность окисления металлов наблюдается в тех случаях, когда продукты окисления не тормозят процесс взаимодействия кислорода с металлом. В частности, такой закономерности подчиняется процесс окисления металлов, образующих порис-тую пленку, или металлов, на которых пленка сильно растрескивается. Так как в последнем случае пленка не препятствует окислению металла ни на каком его этапе, то очевидно, что скорость окисления должна оставаться постоянной в течение всего процесса окисления. В этих условиях скорость химической реакции не зависит от толщины образующейся пленки и описывается уравнением.
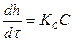 (1)
(1)
где h — толщина образующейся пленки продуктов коррозии металла;
τ - время коррозии;
Кс, —константа скорости химической реакции;
С—концентрация окислителя на поверхности металла, не зависящая от времени его взаимодействия с кислородсодержащей средой (благодаря высокой скорости адсорбции окислителя). После интегрирования уравнения (1) определяем
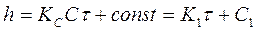 (2)
(2)
В болыпинстве экспериментов были получены малые или равные нулю значения постоянной С1. С учетом этого уравнение (2) принимает вид
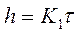 (3);
(3);
Таблица 1. Отношение объемов окислов и израсходованных металлов

Окисные пленки, покрывающие сплошным слоем всю поверхность металла, могут обладать заметными защитными свойствами. Характер образования таких пленок определяется условием сплошности: молекулярный объем окисла V ок должен быть больше объема металла VMе, израсходованного на образование окисла. Из указанного следует, что если V0k/VMе < 1, то пленка не может быть сплошной (рис.1,б). Для расчета отношения V0k/VMе можно пользоваться формулой
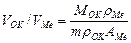 (4)
(4)
где Мок — молекулярная масса соединений; MMе — атомная масса металла; ρок и ρMe — плотность окисла и металла; n — число атомов металла в молекуле окисла.
Все щелочные и щелочноземельные металлы характеризуются отношением V0k/VMе < 1, т. е. не удовлетворяют условию сплошности при окислении их кислородом. В табл. 1 приведены значения отношений объемов некоторых окислов и металлов.
Сплошность пленок окислов является необходимым, но еще недостаточным условием защиты металлов от дальнейшего окисления. При Івозникновении внутренних напряжений пленка окислов может разрушаться частично или полностью. В этом случае нарушается ее сплошность и значительно уменьшаются защитные свойcтва. У пленок с V0k/VMе > 1, отсутствуют высокие защитные свойства. К числу таких окислов относятся Мо03 и VО3. Максимальное значение отношения V0k/VMе равно 2,5. Таким образом, достаточно хорошими защитными свойствами обладают пленки на металлах при 2,5 > V0k/VMе > 1.
При низких температурах (на меди в кислороде до 100° С, на тантале до 150° С, на алюминии, железе, никеле, цинке до 300°С), а также на первых стадиях окисления и при высоких температурах рост тонких окисных пленок сопровождается большим самоторможением во времени, которому отвечает логарифмический закон:
H= Klg(a F+1) (5)
где К и а — постоянные величины.
С утолщением образующихся при высокотемпературном окислении металлсю пленок перемещение через них окислителей в большинстве случаев осуществляется диффузией, которая и контролирует процесс окисления металлов. Скорость установившегося стационарного режима процесса определяется из уравнения
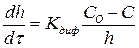 (6)
(6)
где С — концентрация кислорода на внутренней поверхности окисной пленки.
Если на внутренней поверхности пленки не происходит накопления кислорода, т. е. С»0, то уравнение (6) принимает вид
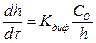 (7)
(7)
После интегрирования уравнения (7) и некоторых преобразований получаем
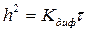 (8)
(8)
Уравнение (8) отображает параболический закон роста пленки. На рис.2 показано увеличение массы, связанное с окислением железа, при различных температурах.
Толщина пленок продуктов коррозии может изменяться в довольно широких пределах и разделяется на три группы: тонкие (невидимые) — до 40 нм; средние (дающие цвета побежалости) — от 40 до 50 нм; толстые (видимые) — больше 500 нм (окалина на стали).
Рассмотренные выше процессы окисления металлов относятся к простейшему случаю образования на металле пленки, состоящей из одного окисла. Вместе с тем при окислении некоторых металлов образуется многослойная окисная пленка. Так, при температурах выше 570°С на железе и углеродистых сталях образуется пленка, состоящая из трех окислов (рис. 3) вюстита FеО, магнетита Fе3О4 и гематита Fе2О3. Со стороны металлической поверхности располагается окисел FеО с небольшим содержанием кислорода, а со стороны окисляющей среды — Fе2О3 с наибольшим содержанием кислорода.
Вюстит представляет собой раствор типа вычитания, когда отдельные узлы не замещены и в кристаллической решетке окисла образуются «дырки». Для сохранения электронейтральности кристалла из ячейки одновременно с ионом должны выйти два электрона, которые появляются в результате перехода двухвалентных ио нов в трехвалентные.
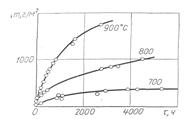
Рис. 2. Увеличение массы, связанное с окислением железа при разных температурах.
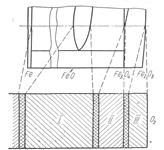
Рис. 3. Структура железной окалины и ее соответствие с диаграммой состояния железо—кислород:
I— закисная фаза; II— магнетит;
III — гематит,
Каждой образовавшейся «дырке» отвечает превращение 2Fе2+ —2Fе3++2е~, где е • — электрон.
Магнетит образует сплошную кубическую решетку типа шпинели (минерал состава Мg0-А12О3). Элементарная ячейка магнетита состоит из 8Fе2+ + 16Fе3++32О2.
Фаза α - гематит имеет сплошную ромбоэдрическую решетку типа корунда Аl2О3. Фаза γ-гематит неустойчива при высоких температурах и сравнительно легко переходит в со Fе2О3. Первая из них имеет почти такую же кристаллическую решетку, как магнетит. В табл. 2 показана схема процесса образования окалины при окислении стали.
Таблица 2. Схема процесса образования окалины при окислении
|
|
|
|
|
Дата добавления: 2014-01-05; Просмотров: 767; Нарушение авторских прав?; Мы поможем в написании вашей работы!