КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Ориентация и реакционная способность 1 страница
Если бензольное кольцо уже содержит заместитель, то:
1.
1. реакция может протекать быстрее или медленнее, чем с самим бензолом;
2. возможно образование трех разных продуктов замещения
Влияние имеющегося в бензольном кольце заместителя можно объяснить исходя из его электронных эффектов. По этому признаку заместители можно разделить на 3 основных группы:
1. Заместители, ускоряющие реакцию по сравнению с незамещенным бензолом (активирующие) и направляющие замещение в орто,- пара- положения.
2. Заместители, замедляющие реакцию (дезактивирующие) и направляющие замещение в орто,-пара- положения.
3. Заместители, замедляющие реакцию (дезактивирующие) и направляющие замещение в мета - положения.
Заместители, отмеченные в п.п. 1,2 (орто-,пара-ориентанты) называются заместителями I-го рода; отмеченные в п.3 (мета-ориентанты) - заместителями II-го рода. Ниже приведено отнесение обычно встречающихся заместителей в соотвествие с их электронными эффектами. Очевидно, что электрофильное замещение будет происходить тем быстрее, чем более электронодонорным является заместитель в ядре, и тем медленнее, чем более электроноакцепторным является заместитель в ядре.
7.р, π- Сопряжение в органических молекулах.
pn-Сопряжение. Этот вид сопряжения чаще всего проявляется в соединениях, содержащих структурный фрагмент -CH=CH-X, где X - гетероатом, имеющий неподеленную пару электронов (прежде всего O или N). К ним относятся, например, виниловые эфиры, в молекулах которых осуществляется сопряжение двойной связи с р -орбиталью атома кислорода. Делокализованная трехцен- тровая связь образуется путем перекрывания двух р-АО sр2-гиб- ридизованных атомов углерода и одной р -АО гетероатома с парой и-электронов.
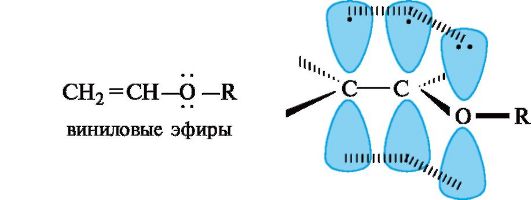
Образование аналогичной делокализованной трехцентровой связи имеется в карбоксильной группе. Здесь в сопряжении участвуют π-электроны связи С=О и n-электроны атома кислорода группы ОН. К сопряженным системам с полностью выровненными связями и зарядами относятся отрицательно заряженные частицы, например ацетат-ион.

Направление смещения электронной плотности обозначается изогнутой стрелкой.
Существуют и другие графические способы отображения результатов сопряжения. Так, структура ацетат-иона (I) предполагает, что заряд равномерно распределен по обоим атомам кислорода (как показано на рис. 2.7, что соответствует действительности).
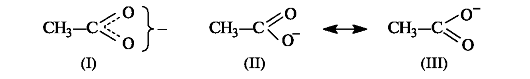
Структуры (II) и (III) применяются в теории резонанса. Согласно этой теории реальная молекула или частица описывается набором определенных так называемых резонансных структур, которые отличаются друг от друга только распределением электронов. В сопряженных системах основной вклад в резонансный гибрид вносят структуры с различным распределением π-электронной плотности (двусторонняя стрелка, связывающая эти структуры, является специальным символом теории резонанса).
Предельные (граничные) структуры в действительности не существуют. Однако они в той или иной степени «вносят вклад» в реальное распределение электронной плотности в молекуле (частице), которую представляют в виде резонансного гибрида, получающегося путем наложения (суперпозиции) предельных структур.
В ρ,π-сопряженных системах с уг- леродной цепью сопряжение может осуществляться при наличии рядом с π-связью атома углерода с негибридизованной р-орбиталью. Такими системами могут быть промежуточные частицы - карбанионы, карбокатионы, свободные радикалы, например, аллильной структуры. Свободнорадикальные аллильные фрагменты играют важную роль в процессах пероксидого окисления липидов.
В аллил-анионе CH2=CH-CH2 sр2-гибридизованный атом углерода С-3 поставляет в общую сопряженную
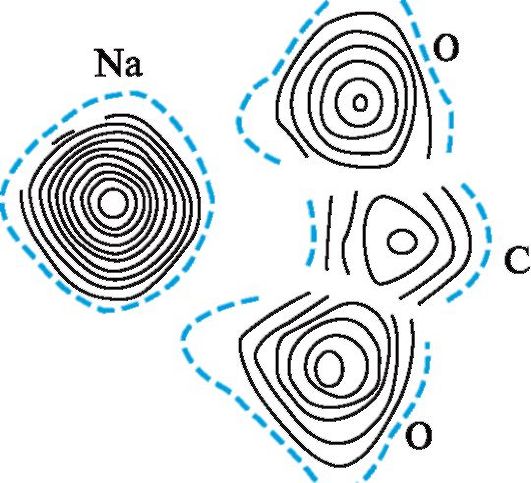
Рис. 2.7. Карта электронной плотности группы COONa в пе- нициллине
систему два электрона, в аллильном радикале CH2=CH-CH2+ - один, а в аллильном карбокатионе CH2 =CH-CH2+ не поставляет ни одного. В результате при перекрывании p-АО трех sp2-гибридизованных атомов углерода образуется делокализованная трехцентровая связь, содержащая четыре (в карбанионе), три (в свободном радикале) и два (в карбокатионе) электрона соответственно.
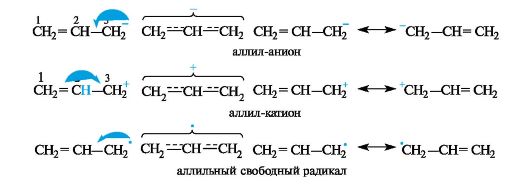
Формально атом С-3 в аллил-катионе несет положительный заряд, в аллильном радикале - неспаренный электрон, а в аллил-анионе - отрицательный заряд. В действительности в таких сопряженных системах имеется делокализация (рассредоточение) электронной плотности, что приводит к выравниванию связей и зарядов. Атомы С-1 и С-3 в этих системах равноценны. Например, в аллил-катионе каждый из них несет положительный заряд +1/2 и связан «полуторной» связью с атомом С-2.
Таким образом, сопряжение приводит к существенному различию в распределении электронной плотности в реальных структурах по сравнению со структурами, изображаемыми обычными формулами строения.
8.Общие закономерности реакций нуклеофильного замещения.
Реакции нуклеофильного замещения атомов галогенов в ароматических соединениях широко используются в органической химии для получения различных практически важных соединений. Однако возможности этих реакций применительно к ароматическим галогенпроизводным, не содержащим сильных электроноакцепторных.заместителей, существенно ограничиваются их относительно малой реакционной способностью.
Классический способ активации ароматического кольца по отношению к действию нуклеофильных реагентов заключается во введении в орто- и пара-псшожения по отношению к атогду галогена электроноакцепторных заместителей, среди которых наиболее предпочтительным с точки зрения легкости введения и эффективности влияния является нитрогруппа. Такой способ активации сужает сферу синтетического использования реакций ароматического нуклеофильного замещения. Несмотря на то, что в случае не активированных этим способом соединений трудность протекания реакций указанного типа частично пре-одалевается благодаря использованию катализаторов, в первую очередь соединений меди/l/, а также никеля/2/ и палладия/3/, проблема поиска новых путей активации ароматического кольца по отношению к действию нуклеофильных агентов продолжает оставаться актуальной.
В этой связи обращает на себя внимание явление, обнаруженное при изучении химии ij-ареновых комплексов переходных металлов и заключающееся в том, что в результате ^-координации ароматического кольца с атомами хрома/4/, железа/5/ или марганца/6/ резко облегчаются процессы нуклеофильного замещения атомов галогенов. Влияние такой координации на скорость протекания этих реакций весьма велико и сравнимо с эффектом одной/4/ или двух/5/ орто- пара-расположенных нитрогрупп. Следует отметить, что этот способ активации ароматического кольца принципиально отличается от введения электроноакцепторных заместителей, поскольку он не, связан с замещением атомов водорода ароштического лиганда, а процессы комплекс ообра-зования ароматических соединений с металлами и обратные процессы выведения свободного ароматического соединения по крайней мере для некоторых типов комплексов протекают достаточно легко. Б связи с этим можно надеяться на то, что рассматриваемые свойства 1|-арено-вых комплексов металлов могут существенно расширить синтетические возможности, связанные с реакциями ароматического нуклеофильного замещения. Такая перспектива предопределяет целесообразность углубленного изучения механизма реакций "Ц-ареновых комплексов металлов с нуклеофильными агентами.
С этой точки зрения наиболее интересными представляются т^-аре-новые комплексы хрома двух типов - арентрикарбонильные и бисарено-вые. Во-первых, эти комплексы являются, вероятно, наиболее доступными моделями, на основе которых можно изучить влияние целого ряда факторов на подвижность атома галогена в 7| -координированном арено-вом кольце. Во-вторых, легкость получения аренхромтрикарбонильных комплексов и выведения ароматического соединения из координации с фрагментом Сг (С0)3, а также высокая скорость нуклеофильного замещения атома галогена позволяет использовать эти комплексы в органическом синтезе. Полученные данные о закономерностях протекания реакций нуклеофильного замещения атома галогена в ареновых комплексах хрома, можно надеяться, будут полезными также и для углубления понимания природы связи между ареновыми лигандами и атомом хрома.
В соответствии с этим настоящая работа посвящена исследованию закономерностей нуклеофильного замещения атома галогена в ароматических соединениях, координированных с хроме од ержащими металло-комплексными фрагментами, выявлению эффектов указанной координации на нуклеофильную подвижность атомов галогенов, а также на характер влияния заместителей в ароматическом соединении и развитию на этой основе экспериментально обоснованных представлений о влиянии координации на механизм реакций ароматического нуклеофилъного замещения.
В результате проведения кинетических исследований было найдено, что реакции нуклеофилъного замещения атома галогена в бисареновых и арентрикарбонильных комплексах хрома при действии метилата натрия имеют общий второй порядок (первый как по субстрату, так и по реагенту) и активационные параметры, характерные для реакций нуклеофилъного замещения атома галогена в активированных сильными электроноакцепторными заместителями некоординированных аренах. В то же время было обнаружено, что реакция нуклеофилъного замещения атома хлора в 0|-пара-дихлорбензол)хромтрикарбониле и бис
•хлорбензол)хроме(1+) при действии пиперидина имеет третий порядок (первый по субстрату и второй по реагенту), что обычно интерпретируется как наличие катализа пиперидином. Полученные значения активационных параметров для этой реакции в случае бис-(ц-хл орбенз ал) хрома (1+) характерны для катализируемых реагентом реакций некоординированных арилгалогенидов с пиперидином. В результате проведенных исследований получено большее экспериментальное обоснование сформулированного ранее предположения о протекании реакций нуклеофильного замещения атома галогена в аредовых лиган-дах комплексов переходных металлов по двухстадийному механизму с промежуточным образованием (5-комплекса, кал и в случае некоординированных активированных галогенбензолов.
9.Номенклатра органических соединений.
Это первый вопрос
10. Нуклеиновые кислоты.
Нуклеи́новая кисло́та (от лат. nucleus — ядро) — высокомолекулярное органическое соединение, биополимер (полинуклеотид), образованный остаткаминуклеотидов. Нуклеиновые кислоты ДНК и РНК присутствуют в клетках всех живых организмов и выполняют важнейшие функции по хранению, передаче и реализациинаследственной информации.
Описаны многочисленные методики выделения нуклеиновых кислот из природных источников. Основными требованиями, предъявляемыми к методу выделения, являются эффективное отделения нуклеиновых кислот от белков, а также минимальная степень фрагментации полученных препаратов. Классический метод выделения ДНК был описан в 1952 году и используется в настоящее время без значительных изменений[7]. Клеточные стенки исследуемого биологического материала разрушаются одним из стандартных методов, а затем обрабатываются анионным детергентом. При этом белки выпадают в осадок, а нуклеиновые кислоты остаются в водном растворе. ДНК может быть осаждена в виде геля осторожным добавлением этанола к её солевому раствору. Концентрацию полученной нуклеиновой кислоты, а также наличие примесей (белки, фенол) обычно определяют спектрофотометрически по поглощению на А260 нм.
Нуклеиновые кислоты легко деградируют под действием особого класса ферментов — нуклеаз. В связи с этим при их выделении важно обработать лабораторное оборудование и материалы соответствующими ингибиторами. Так, например, при выделенииРНК широко используется такой ингибитор рибонуклеаз как DEPC.
Физические свойства
Нуклеиновые кислоты хорошо растворимы в воде, практически не растворимы в органических растворителях. Очень чувствительны к действию температуры и критическим значениям уровня pH. Молекулы ДНК с высокой молекулярной массой, выделенные из природных источников, способны фрагментироваться под действием механических сил, например при перемешивании раствора. Нуклеиновые кислоты фрагментируются ферментами — нуклеазами.
Строение
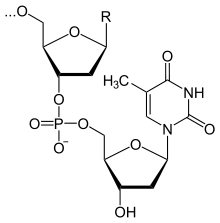

Фрагмент полимерной цепочки ДНК
Полимерные формы нуклеиновых кислот называют полинуклеотидами. Цепочки из нуклеотидов соединяются через остаток фосфорной кислоты (фосфодиэфирная связь). Поскольку в нуклеотидах существует только два типа гетероциклических молекул, рибоза и дезоксирибоза, то и имеется лишь два вида нуклеиновых кислот — дезоксирибонуклеиновая (ДНК) и рибонуклеиновая(РНК).
Мономерные формы также встречаются в клетках и играют важную роль в процессах передачи сигналов или запасании энергии. Наиболее известный мономер РНК — АТФ, аденозинтрифосфорная кислота, важнейший аккумулятор энергии в клетке.
ДНК и РНК
· ДНК (дезоксирибонуклеиновая кислота). Сахар — дезоксирибоза, азотистые основания: пуриновые — гуанин (G), аденин (A),пиримидиновые — тимин (T) и цитозин (C). ДНК часто состоит из двух полинуклеотидных цепей, направленных антипараллельно.
· РНК (рибонуклеиновая кислота). Сахар — рибоза, азотистые основания: пуриновые — гуанин (G), аденин (A), пиримидиновыеурацил (U) и цитозин (C). Структура полинуклеотидной цепочки аналогична таковой в ДНК. Из-за особенностей рибозы молекулы РНК часто имеют различные вторичные и третичные структуры, образуя комплементарные участки между разными цепями.
11.Донорно-акцепторные связи в органических молекулах.
Донорно-акцепторная связь
Донорно-акцепторная связь (координационная связь, семиполярная связь) — химическая связь между двумя атомами или группой атомов, осуществляемая за счет неподеленной пары электронов одного атома (донора) и свободного уровня другого атома (акцептора). Донорно-акцепторная связь образуется часто при комплексообразовании за счет свободной пары электронов, принадлежавшей (до образования связи) только одному атому (донору). Донорно-акцепторная связь отличается от обычной ковалентной только происхождением связевых электронов. Реакция аммиака с кислотой состоит в присоединении протона, отдаваемого кислотой, к неподеленным электронам азота:
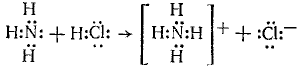
В ионе NH4+ все четыре связи азота с водородом равноценны, хотя отличаются происхождением. Донорами могут быть атомы азота, кислорода, фосфора, серы и др. Роль акцепторов могут выполнять протон, а также атомы с незаполненным октетом, напр. атомы элементов III группы таблицы Д. И. Менделеева, а также атомы-комплексообразователи, имеющие незаполненные энергетические ячейки в валентном электронном слое. Донорно-акцепторная связь называется иначе семиполярной (полуполярной), так как на атоме-доноре возникает эффективный положительный заряд, а на атоме-акцепторе — эффективный отрицательный заряд. Изображают эту связь стрелкой, направленной от донора к акцептору.
В процессе образования ковалентной связи связанные электроны проникают во внешнюю оболочку обоих связываемых атомов. Соответственно, количество электронов на внешних оболочках этих атомов увеличивается на один электрон.
Существует и другой механизм образования связи: за счет пары электронов только ОДНОГО из взаимодействующих атомов.
В результате образования молекулы атом Х может дополнить свою оболочку до 8 электронов, однако в образовании связи будут принимать участие не все 8 электронов, а только часть. Электроны, не принимающие участия в образовании химических связей, считаются свободными, несвязанными.
Атом Y может отдать все свои электроны для образования связей, однако в результате обобществления электронов с другим атомом иметь на внешней оболочке менее 8 электронов.
Атомы со свободными (несвязанными) электронами являются донорами электронов при образовании донорно-акцепторной химической связи. Их партнерами, в первую очередь, становятся это те атомы, чьи оболочки содержат менее 8 электронов. Это атомы в молекулах, образованных элементами 2-го и 3-го периодов (с числом электронов во внешнем слое менее 4).
Атомы натрия (Na), магния (Mg) и алюминия (Al) после образования максимального числа ковалентных связей, к примеру, после образования молекул NaF, MgF2, AlF3 содержат соответственно 2, 4 и 6 электронов во внешней оболочке. Внешние оболочки атомов Na, Mg и Al имеют менее 8 электронов, т.е. остаются ненасыщенными.
Атомы азота (N), кислорода (O) и фтора (F) в соединениях аммоний (NH3), вода (H2O) и фторид водорода (HF) содержат соответственно 2, 4 и 6 электронов во внешних оболочках, которые не принимают участия в образовании химических связей, т.е. их считают свободными. Поскольку в образованных соединениях NH3, H2O и HF во внешних оболочках атомов N, O и F находится 8 электронов, свободные, несвязанные электроны не могут участвовать в образовании ковалентной связи из-за того, что внешняя оболочка этих атомов насыщена электронами.
Хорошо известны следующие устойчивые соединения:
H3B ← NH3; H3B ← N(CH3)2; F3B ←NH3; F3B ←O(CH3)2; Cl2Be ←O(C2H5)2; Cl3Al ←NH3; и др.
Стрелочки (←) здесь обозначают связи донор-акцептор.
Доноры (атомы N и O) в этих примерах отдают 2 электрона. Акцепторами являются B, Be и Al.
При образовании донорно-акцепторнойсвязи атомы-акцепторы увеличивают число электронов в своих внешних оболочках на 2.
12.Бимолекулярное нуклеофильное замещение.
Бимолекулярное нуклеофильное замещение (SN2) включает одновременное перемещение электрона от замещающего агента к алкильной группе и от последней к отщепляемой группе. Как правило, эти перемещения электронов не полностью сбалансированы в переходном состоянии, в связи с чем можно ожидать влияния эффекта на скорость реакции. Роль этого эффекта невелика, так как он зависит только от неполного сбалансирования перемещения электронов; для определения направленности этого эффекта необходим детальный анализ. Как будет показано в разд
Бимолекулярное нуклеофильное замещение - реакция нуклеофильного замещения у алифатического атома углерода, в скоростьлимитирующей стадии которой участвуют две частицы. Протекает как согласованный одностадийный процесс
Процесс бимолекулярного нуклеофильного замещения (SV2) протекает с ярко выраженным обращением конфигурации.
Механизм бимолекулярного нуклеофильного замещения у фосфора был изучен Достровским и Халлманом [36], которым не удалось обнаружить какого бы то ни было обмена изотопов кислорода между (С2Н50) 2Р018С1 и обычной водой, или между (С2Н50) 2РОС1 и Н2018 в процессе прерываемого гидролиза. Эти результаты исключают механизм с быстрым обратимым образованием промежуточного соединения и, согласно этим авторам, заставляют считать возможным, ч-о реакция представляет собой одностадийное синхронное ЧЯМРПТРНМА Этот опыт не исключает, однако, возможность механизма, включающего образование промежуточного комплекса, при котором вторая стадия протекает гораздо быстрее, чем реакция, обратная первой.
Механизм бимолекулярного нуклеофильного замещения у фосфора был изучен Достровским и Халлманом [36], которым не удалось обнаружить какого бы то ни было обмена изотопов кислорода между (С2Н5О) 2РО18С1 и обычной водой, или между (С2Н50) 2РОС1 и Н2О18 в процессе прерываемого гидролиза. Эти результаты исключают механизм с быстрым обратимым образованием промежуточного соединения и, согласно этим авторам, заставляют считать возможным, ч-о реакция представляет собой одностадийное синхронное замещение. Этот опыт не исключает, однако, возможность механизма, включающего образование промежуточного комплекса, при котором вторая стадия протекает гораздо быстрее, чем реакция, обратная первой.
Для обычного бимолекулярного нуклеофильного замещения при атоме углерода установлен следующий ряд скоростей реакции: первичный вторичный третичный. Реакции раскрытия цикла окиси (формула XVIII) протекают в соответствии с этими данными.
При бимолекулярном нуклеофильном замещении разрыв старой и образование новой связи происходят одновременно, поэтому оба компонента реакции участвуют в стадии, определяющей скорость реакции.
В бимолекулярном нуклеофильном замещении электроноакцепторные заместители, особенно заместители, сопряженные с ароматической системой, такие, как нитриль-ная, карбонильная, циан - или сульфонильная группа, облегчают атаку реагента; гетероатомы в положении 2 или 4 в пиридине действуют подобным же образом.
Далее следует бимолекулярное нуклеофильное замещение 5 2 группы X гидрид-ионом при участии второго гидридного эквивалента, причем образуется алкоголят II. Если скорость реакции AdN значительно превышает скорость реакции 5л - 2, в реакционной смеси накапливается алкоголят I, гидролиз которого приводит к альдегиду. Это условие выполняется при восстановлении амидов кислот в тех случаях, когда нуклеофильность атома азота понижена, например, за счет сопряжения с ароматическим ядром.
По механизму бимолекулярного нуклеофильного замещения (5 2) протекают реакции метанола и большинства пространственно незатрудненных первичных спиртов, из которых карбкатионы образуются с трудом.
Такие превращения путем бимолекулярного нуклеофильного замещения, вероятно, являются общим свойством нитрозилышх соединений.
Как известно, бимолекулярное нуклеофильное замещение (5Лт2) всегда протекает с вальденовским обращением. Основываясь на изложенном выше материале по стереохимии и кинетике, мы могли констатировать [68, 71], что бимолекулярное электрофильное замещение у насыщенного атома углерода SE2 протекает с сохранением стереохимической конфигурации.
Последняя стадия представляет собой обычное бимолекулярное нуклеофильное замещение, движущей силой которого является образование стабильного трифенилфосфиноксида.
Последняя стадия представляет собой обычное бимолекулярное нуклеофильное замещение, движущей силой которого является образование стабильного трифенилфосфиноксида. Для интермедиата Р1гзР (Ж СГ обычно принято строение фосфоиневой соли с хлорид - ионом в качестве протиаоиоиа. Однако во многих случаях более обоснованной является структура ковалентного фосфораиа Ph3P (Cl) - OR с пятикоординированным атомом фосфора. Такая структура доказана для пространственно затрудненных спиртов - неопентилового, эндо - и экзо-норборнеолов, холестерина и др. В любом случае превращение спирта в алкилгалогенид протекает с обращением конфигурации.
В случае реакций бимолекулярного нуклеофильного замещения при насыщенном углеродном атоме качественная картина полярного влияния заместителей на скорость процесса, как правило, довольно ясна, если варьирование полярного эффекта производится в нукдео-филе или уходяцеи группе. Обозначение SN2 расшифровывается как бимолекулярное нуклеофильное замещение.
Примерами таких реакций являются бимолекулярное нуклеофильное замещение и отщепление, а также реакции переноса протона.
Ранее эта обычная реакция бимолекулярного нуклеофильного замещения имела крайне ограниченную область применения из-за низкого выхода сложных эфиров в протонных растворителях (вода, спирты), где карбоксилат-ион сильно сольватирован с помощью водородной связи и поэтому обладает низкой реакционной способностью.
Нами рассмотрены основные черты бимолекулярного нуклеофильного замещения.
Как и в других случаях бимолекулярного нуклеофильного замещения, разветвление углеродной цепи в р-положении к одиночному атому хлора затрудняет его гидролитическое отщепление: хлорид (СН3ОСН2) 3 - - ССН2С1 устойчив к щелочному гидролизу настолько, что почти не изменяется после 150 час.
Естественно, что в случае бимолекулярного нуклеофильного замещения в аналогичных системах наблюдается обратная зависимость при сравнении влияния заместителей на скорость реакции.
Естественно, что в случае бимолекулярного нуклеофильного замещения в аналогичных системах при сравнении влияния заместителей на скорость реакции наблюдается обратная зависимость.
Еще более прочная связь между бимолекулярным нуклеофильным замещением и инверсией установлена Хьюзом и сотрудниками в 1935 г. Они изучили параллельно рацемизацию 2-иодоктана при действии иодид-ионов и накопление в 2-иодоктане радиоактивного иода в результате взаимодействия с радиоактивными иодид-ионами.
При взаимодействии с кетоном в результате бимолекулярного нуклеофильного замещения образуется алкоголят-ион, который немедленно соединяется с нейтральным гидридом алюминия, образуя новый ион.
Эти реакции вполне правдоподобно были представлены как бимолекулярное нуклеофильное замещение молекулами воды у атома углерода, а не у четырех-лигандного фосфора, происходящее в одной или другой ионной форме кислоты, сопряженной монометиловому эфиру.
Реакция цианирования феррициниевых солей не является реакцией бимолекулярного нуклеофильного замещения у атома углерода пятичленного кольца.
13.Стереоизомерия органических соединений.
Пространственные изомеры — это соединения, имеющие одинаковую молекулярную формулу, одинаковый порядок связывания атомов углерода в молекулах, но отличающиеся друг от друга расположением атомов в пространстве.
Пространственные изомеры называют также стереоизомерами (от греч. stereos — пространственный).
Пространственная изомерия подразделяется на конфигурационную и конформационную.
Но прежде чем перейти к рассмотрению этих видов стерео-изомерии, остановимся на способах изображения пространственного строения молекул органических соединений.
Для изображения пространственного строения молекул, их конфигураций или конформаций на плоскости применяют специальные стереохимические формулы.
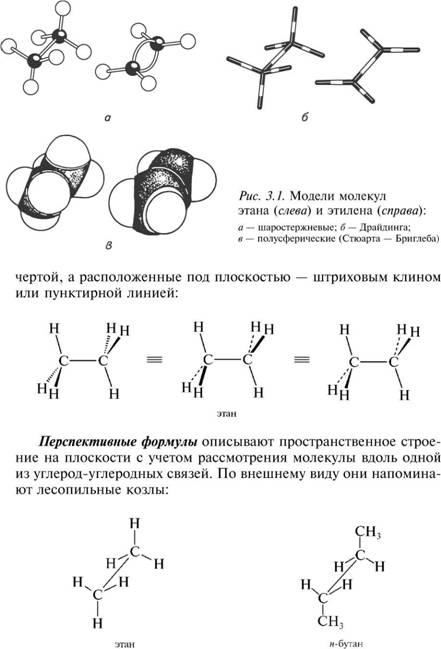
Для наглядного представления молекул как органических, так и неорганических соединений используют молекулярные модели, которые дают возможность судить о порядке связывания и взаимном расположении атомов в молекуле.
Наиболее часто используют три основных типа моделей: ша-ростержневые (модели Кекуле—Вант-Гоффа), скелетные (модели Драйдинга) и полусферические (модели Стюарта—Бриглеба).
Модели позволяют судить не только о взаимном расположении атомов в молекуле; они также удобны для рассмотрения валентных углов и возможности вращения вокруг простых связей. Модели Драйдинга учитывают межатомные расстояния, а модели Стюарта—Бриглеба отражают также и размеры атомов.
Ниже, на рис. 3.1, приведены модели молекул этана и этилена.
этан этилен
Стереохимические формулы. Для изображения пространственного строения молекулы на плоскости чаще всего используют сте-реохимические и перспективные формулы, а также проекционные формулы Ньюмена.
В стереохимических формулах химические связи, расположенные в плоскости чертежа, изображают обычной чертой; связи, находящиеся над плоскостью,— жирным клином или жирной
3. Изомерия органических соединений. Пространственное строение молекул
п^н ни^і^иипин приъкцииппшл, ціирміул АаОпгмъпи тил^А)Л) рассматривают в направлении одной С—С-связи таким образом, чтобы атомы, образующие данную связь, заслоняли друг друга. Из выбранной пары ближний к наблюдателю атом углерода изображают точкой, а дальний — окружностью. Химические связи ближнего атома углерода с другими атомами представляют линиями, берущими начало от точки в центре круга, а дальнего — от окружности:

ириекцииппои^ цмиушулоъ ул^ишъри представляют собой как бы
проекцию молекулы на плоскости.
Рассмотрим построение проекционной формулы Фишера для бутанола-2 СН3—СН(ОН)—СН2—СН3. Для этого модель молекулы располагают таким образом, чтобы атом углерода, связанный с гидроксильной группой, находился в плоскости чертежа, а заместители, расположенные горизонтально, были над плоскостью, расположенные вертикально — за плоскостью чертежа. При проецировании такой модели на плоскость получают проекционную формулу Фишера, в которой связи, находящиеся над плоскостью, изображают горизонтальной линией, а связи, находящиеся за плоскостью,— вертикальной линией. В точке пересечения этих линий находится атом углерода, который обычно не обозначается символом:
|
|
Дата добавления: 2015-04-24; Просмотров: 826; Нарушение авторских прав?; Мы поможем в написании вашей работы!