КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Перинатальная паталогия
|
|
|
|
Гломерулопатии могут быть врожденными и приобретенными. Врожденные гломерулярные заболевания (наиболее часто встречается синдром Альпорта, при котором нефрит сочетается с дегенерацией слухового нерва и катарактой) наблюдаются очень редко. В данной лекции будут рассмотрены приобретенные гломерулярные заболевания.
Причиной развития гломерулонефритов могут быть инфекционные и неинфекционные факторы (антигены), наследственная предрасположенность, лекарственные воздействия, избыточная инсоляция и др.
Патогенетическая классификация включает в себя иммунологическое и неиммунологическое повреждение. Иммунологическое повреждение может антительным, то есть происходить в результате действия антигломерулярных антител (против измененных антигенов собственной базальной мембраны клубочков анти-БМК) или иммунокомплексным, в результате накопления иммунных комплексов, составной частью которых являются антигены этиологического фактора. Антигены, входящие в состав иммунных комплексов, могут образовываться из бактерий, паразитов, лекарств и т.д. Неиммунологические повреждения почек в большинстве случаев являются сопутствующим заболеванием(например, при сахарном диабете). Гистологическая классификация основывается на типе реакции клубочка на повреждение (например, пролиферация, утолщение мембраны и т.д.) Гистологическая картина помогает в определении этиологии и выборе терапевтической тактики.
Ниже приведена комплексная классификация гломерулонефритов, основанная на клинических (врожденный или приобретенный; острый или хронический), морфологических (пролиферативный, мембранозный, с минимальными изменениями) и иммунологических характеристиках (табл. 1)
Таблица 1
Классификация гломерулопатий
| Врожденные гломерулонефриты Наследственный нефрит (синдром Альпорта) Врожденный нефротический синдром Первичные приобретенные гломерулонефриты1 Гломерулонефрит с минимальными изменениями Постинфекционный (постстрептококковый) гломерулонефрит Подострый (полулунный) гломерулонефрит Синдром Гудпасчера Мезангиопролиферативный гломерулонефрит Мембранозный гломерулонефрит Мембранопролиферативный (мезангиокапиллярный) гломерулонефрит Очаговый гломерулосклероз Вторичные приобретенные гломерулонефриты2 Хронический гломерулонефрит Другие гломерулярные заболевания Диабетическая нефропатия Амилоидоз |
1 «Первичные» означает то, что поражение почек является главным проявлением заболевания.
2 «Вторичный» означает то, что поражение почек является частью системного заболевания, например, при системной красной волчанке и прогрессирующем системном склерозе.
ПЕРВИЧНЫЕ ГЛОМЕРУЛЯРНЫЕ ЗАБОЛЕВАНИЯ
Гломерулярная болезнь (гломерулонефрит) с минимальными изменениями
Гломерулярная болезнь с минимальными изменениями (идиопатический нефротический синдром) наиболее часто развивается у детей в возрасте до 8 лет (80%) и редко у взрослых и сопровождается нефротическим синдромом. Пик заболеваемости лежит между 2 и 4 годами. Чаще болеют мальчики. Это заболевание также известно под названием липоидный нефрит, т.к. при нем часто в клетках канальцев обнаруживаются жиры, что является наиболее значимым микроскопическим признаком.
Этиология и патогенез. Основным изменением при данной патологии является снижение в базальной мембране содержания полианионов (в основном гепаран сульфата (протеогликан)), что приводит к снижению отрицательного заряда в мембране. В результате ослабевает фильтрационный барьер для анионных молекул плазмы, например, альбуминов, которые начинают проникать сквозь мембрану в больших количествах. При этом из всех белков плазмы теряются в основном альбумины, поэтому для гломерулярной болезни с минимальными изменениями характерна “селективная” протеинурия. Слияние отростков эпителиальных клеток является неспецифическим ответом на повышенную фильтрацию белков.
Причина химических изменений в мембране неизвестна. У некоторых больных заболевание развивается после респираторных инфекций и профилактических прививок. Иммунологическая основа болезни подтверждается следующими признаками: 1) связью заболевания с инфекциями, иммунизацией и атопическими реакциями, такими как сенная лихорадка и экзема; 2) отличной эффективностью терапии иммуносупрессивными лекарствами, например, кортикостероидами. Так как болезнь нередко развивается при лимфоме Ходжкина, при которой обнаруживается нарушение функции Т-лимфоцитов, некоторые исследователи пришли к мнению, что данная патология развивается в результате действия клеточных иммунологических реакций.
Патологическая анатомия. При световой микроскопии никаких изменений не обнаруживается (отсюда термин “минимальные изменения”). При иммунофлюоресценции также не выявляется отложение иммуноглобулинов и комплемента. При электронной микроскопии обнаруживается слияние отростков подоцитов (“болезнь эпителиальных клеток”) (рис. 3). Эти изменения исчезают во время ремиссии.

Рис. 3. Гломерулонефрит с минимальными изменениями.
Нарушения обнаруживаются только при электронной микроскопии: слияние отростков подоцидов, которое является неспецифическим.
Клинические проявления. Болезнь с минимальными изменениями проявляется нефротическим синдромом. Протеинурия в большинстве случаев является “высоко селективной” с потерей только низкомолекулярных анионных белков. “Селективность” протеинурии измеряется соотношением содержания трансферрина (низкомолекулярный белок) и IgG (высокомолекулярный белок) в моче. При высокоселективной протеинурии это отношение велико, при неселективной протеинурии оно равно приблизительно 1. Гематурия, гипертензия и азотемия отсутствуют. Клинически проявляется нефротическим синдромом, который хорошо лечится кортикостероидами. Прогноз у детей хороший, у взрослых исход может быть неблагоприятным (развитие нефросклероза и ХПН).
Заболевания, связанные с накоплением иммунных комплексов
Повреждения, вызванные иммунными комплексами, приводит к развитию различных реакций в клубочках, которые зависят от природы иммунных комплексов. Это повреждение может проявляться в виде диффузного либо фокального пролиферативного гломерулонефрита.
Гломерулонефриты в настоящее время делят на:
- постинфекционный (острый диффузный пролиферативный);
- быстропрогрессирующий, злокачественный (подострый);
- хронический.
Постинфекционный (острый диффузный пролиферативный) гломерулонефрит является одним из типов повреждения клубочков, имеющий постинфекционную этиологию. Раньше считалось, что заболевание обусловлено инфекцией, возбудителем которой является β-гемолитический стрептококк (постстрептококковый гломерулонефрит). Однако выяснено, что причиной данного заболевания могут быть не только стрептококки (нестрептококковый гломерулонефрит). Существуют сообщения об этиологической роли таких микроорганизмов, как Staphylococcus aureus, Streptococcus pneumoniae, Neisseria meningitidis, плазмодия малярии, Toxoplasma gondii и некоторых вирусов. Иммунные комплексы, формирующиеся между антигенами и антителами в организме хозяина, откладываются на фильтрационной мембране клубочка, происходит фиксация комплемента и развитие воспаления. До сих пор не обнаружен специфический стрептококковый антиген, который участвует в образовании иммунных комплексов.
Только из-за значительного превалирования над другими типами постстрептококковый гломерулонефрит рассматривается в данной лекции самостоятельно.
Острый постстрептококковый гломерулонефрит является одним из самых распространенных заболеваний у детей. У взрослых он встречается намного реже.
Наиболее частой причиной является β-гемолитический стрептококк группы А. Не все стрептококковые инфекции представляют опасность в плане развития гломерулонефрита. Существуют так называемые “нефритогенные” штаммы стрептококков М типа, особенно серотипов 1, 4, 12 и 49.
Первичная инфекция обычно проявляется в виде ангины, фарингита, реже отита и воспалительных поражений кожи. Имеются существенные географические различия в распространенности и тяжести постстрептококкового гломерулонефрита. Например, в Индии он является наиболее частым поражением почек, а в Англии встречается очень редко.
Клинические проявления. У большинства больных постстрептококковый гломерулонефрит начинается внезапно, остро в виде недомогания, повышения температуры и тошноты через 7-14 дней после ангины, с быстрым развитием нефритического синдрома. Появляется олигурия (резкое снижение объема мочи) и моча темнеет (цвета “мясных помоев”) в результате гематурии. При объективном исследовании обнаруживается отеки на лице, особенно периорбитальные, и слабо выраженная гипертензия. В крови определяется повышение титра антистрептолизина О и значительное уменьшение фракции С3 комплемента. Тесты на ЦИК обычно положительные. Повышение содержания в крови азотистых шлаков (мочевой кислоты и креатинина) говорит о нарушении функции почек. В анализах мочи определяется гематурия, увеличивается количество лейкоцитов, появляются цилиндры, а также присутствует различной степени выраженности протеинурия. При исследовании глотки и кожи на микрофлору обычно стрептококк не обнаруживается, т.к. к этому времени инфекция обычно разрешается.
Патологическая анатомия. Макроскопически почки слегка увеличены из-за отека. Капсула снимается легко, обнажая гладкую ровную поверхность. В тяжелых случаях на поверхности могут быть многочисленные петехии (красный крап).
При микроскопическом исследовании определяется диффузный гломерулонефрит. В начальной стадии клубочки увеличены, в них увеличено количество клеток (рис. 4). Увеличение количества клеток возникает в результате пролиферации мезангиальных, эндотелиальных и эпителиальных клеток наружного листка капсулы клубочка, а также различной степени выраженности инфильтрации полиморфноядерными лейкоцитами. Выраженный отек и набухание эндотелия приводит к сужению просвета капилляров.
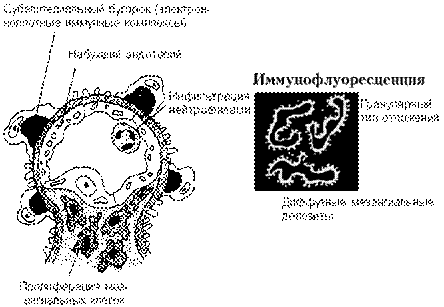
Рис. 4. Постстрептококковый гломерулонефрит.
Постстрептококковый гломерулонефрит характеризуется отложением электронноплотных иммунных комплексов под эпителием и в мезангиуме. Активация комплемента приводит к пролиферации клеток и воспалению.
При световой микроскопии могут обнаруживаться иммунные комплексы в виде характерных “глыбок”, особенно при трихромных окрасках. При электронной микроскопии они представляют собой большие электронно-плотные отложения в виде купола на эпителиальной стороне базальной мембраны (субэпителиальные «горбы») (рис. 4). Также часто определяются отложения иммунных комплексов в мезангиуме, в субэндотелиальных и внутримембранных регионах. При иммунофлюоресценции определяются гранулярные отложения IgG и С3 вдоль базальной мембраны клубочка и в мезангиуме.
Прогноз. Благодаря применению противовоспалительной и иммуносупрессивной терапии 95% пациентов клинически выздоравливают в течение 6 недель. Нормальная функция почек восстанавливается в течение года. Иногда может сохраняться увеличение числа клеток в мезангиуме в течение многих месяцев, и даже лет. Нарушения в осадке мочи могут существовать в течение нескольких лет. У небольшого числа больных отмечается прогрессия с быстрым развитием почечной недостаточности в течение 1-2 лет. У этих больных обнаруживается наличие множественных полулуний (подострый гломерулонефрит – см. ниже).
Прогноз в отдаленные сроки остается пока дискуссионным вопросом. По результатам одних исследований у детей прогноз хороший. Более плохой прогноз определяется у взрослых, у которых ХПН развивается в 30-50% случаев. Однако, следует оговорить, что, возможно, это случаи острого начала хронического гломерулонефрита.
Быстропрогрессирующий (подострый) гломерулонефрит – это редкое заболевание, которое отражает тяжелое поражение клубочков. Подострый гломерулонефрит характеризуется:
- наличием более чем в 70% клубочков эпителиальных полулуний. Полулуния, заполняющие капсулу Боумена, состоят из эпителиальных клеток и макрофагов, пролиферирующих в ответ на выпот фибрина из поврежденных клубочков. Полулуния представляют собой необратимое повреждение клубочка, приводящее к его склерозированию. В полулуниях обязательно находят фибрин, который, как предполагается, и является причиной их образования.
- быстрой прогрессией, приводящей к почечной недостаточности в течение нескольких месяцев. Быстро развивается нефросклероз и гипертензия.
Быстропрогрессирующий гломерулонефрит обычно является конечной стадией многих заболеваний почек, которые приводят к тяжелому повреждению клубочкового аппарата почек (табл. 2). Наиболее часто он развивается после стрептококкового гломерулонефрита и при синдроме Гудпасчера. По патогенезу он одновременно является и иммунокомплексным, и антительным.
Таблица 2
Причины развития быстропрогрессирующего гломерулонефрита
| Постинфекционный Постстрептококковый гломерулонефрит Нестрептококковый гломерулонефрит Инфекционный эндокардит Полиорганные заболевания Синдром Гудпасчера Системная красная волчанка Пурпура Шенляйн-Геноха Болезнь Берже (IgA-нефропатия) Узелковый периартериит Гранулематоз Вегенера Мембранопролиферативный гломерулонефрит Лекарства:пеницилламин Идиопатический Тип I: с анти-БМК антителами (20%) Тип II: с иммунными комплексами (30%) Тип III: иммунонеактивный (50%) |
Существует также группа больных с идиопатическим развитием подострого гломерулонефрита. В этой группе приблизительно у 20% больных в сыворотке были обнаружены анти-БМК антитела и линейный тип отложений при иммунофлюоресценции (тип I), у 30% имели место признаки иммунокомплексного поражения (тип II). У остальных 50% больных при иммунофлюоресценции не было найдено выраженных изменений (иммунонеактивный, или тип III).
Патологическая анатомия. Макроскопически почки резко увеличены (массой до 300-500 г, в норме 120-150 г), дряблые, корковый слой утолщен, набухший, бледный, белого цвета (“большая белая почка”), либо желто-серый, тусклый, с красным крапом и хорошо отграничен от темно-красного мозгового вещества почки (“большая пестрая почка”), либо красный и сливается с полнокровными пирамидами (“большая красная почка”).
Для постановки диагноза необходимо наличие не менее 70% клубочков с полулуниями, т.к. полулуния в небольшом количестве могут обнаруживаться при различных гломерулопатиях. Выражен тубуло-интерстициальный компонент (дистрофические изменения эпителия канальцев, воспалительные инфильтраты в строме).
Лечение обычно малоэффективное и прогноз очень плохой без диализа или трансплантации. Имеются сообщения о развитии подострого гломерулонефрита в трансплантированной почке.
Синдром Гудпасчера встречается редко. Наиболее часто он развивается у подростков, у мужчин несколько чаще, чем у женщин. У 80% больных определяется HLA-DR2 антиген. В сыворотке определяются анти-БМК антитела, направленные против гликопротеиновых антигенов неколлагеновой части коллагена IV типа. Эти антитела обладают способностью реагировать как с базальной мембраной клубочков почек, так и с базальной мембраной альвеол легких.
Патологическая анатомия. Первоначально при световой микроскопии обнаруживается фокальный (очаговый) пролиферативный гломерулонефрит. Затем развивается диффузный гломерулонефрит с формированием эпителиальных полулуний. На поздних стадиях определяется выраженный склероз.
При иммунофлюоресценции определяется накопление IgG и С3 с характерным линейным типом расположения вдоль базальной мембраны. Для постановки диагноза важно обнаружение С3, т.к. линейное накопление IgG вдоль мембраны может наблюдаться и при некоторых неспецифических поражениях, например, при сахарном диабете. Такой же тип накопления IgG и С3 определяется и в альвеолах легких. При электронной микроскопии обнаруживается диффузное и неравномерное утолщение базальной мембраны клубочка, однако, эти проявления являются неспецифическими. У большинства больных в легких обнаруживается обширное повреждение альвеол и внутриальвеолярные кровоизлияния с наличием в их просвете макрофагов с гранулами гемосидерина в цитоплазме.
Клинические проявления. При синдроме Гудпасчера в моче определяется гематурия и протеинурия, которые являются проявлениями быстро прогрессирующего гломерулонефрита. У больных с поражением легких наблюдаются повторные кровохарканья, кашель, при рентгенологическом исследовании – билатеральная инфильтрация легочной ткани. У некоторых больных определяются признаки поражения только почек или только легких. В результате потери крови из-за повторного кровохаркания и гематурии у больных часто развивается тяжелая железодефицитная анемия.
Лечение при синдроме Гудпасчера обычно малоэффективное и прогноз плохой. В большинстве случаев почечная недостаточность развивается в течение года после постановки диагноза.
ХРОНИЧЕСКИЙ ГЛОМЕРУЛОНЕФРИТ
Хронический гломерулонефрит – это заболевание с первичным воспалением клубочков и возможным вторичным вовлечением канальцев и стромы. У большинства больных в анамнезе есть указания на хронические почечные заболевания, однако, у 20% больных даже при первом обращении может быть обнаружена терминальная стадия хронической почечной недостаточности.
Хронические гломерулонефриты делятся на:
- мезангиопролиферативный;
- мембранозный;
- мембранопролиферативный или мезангиокапиллярный.
Мезангиопролиферативный гломерулонефрит. Пролиферация мезангиальных клеток как самостоятельная патология при исследовании биопсий является неспецифической. Эта пролиферация часто носит одновременно фокальный и сегментарный характер. Патогенез во многих случаях неизвестен. При световой микроскопии определяется увеличение количества мезангиальных клеток (более трех ядер в одной дольке). При ШИК-реакции и электронной микроскопии обнаруживаются увеличение количества мезангиального матрикса и электронно-плотные отложения в нем.
Мезангиопролиферативный гломерулонефрит классифицируют на основе доминирующего иммуноглобулина, накапливающегося в клубочке. Наиболее часто в мезангиуме накапливается IgG и С3. Накопление IgA наблюдается при пурпуре Шенляйн-Геноха, системном васкулите с вовлечением почек. Известно всего лишь одно заболевание, имеющее фокальный тип поражения почек, при котором почки поражаются первично – IgA нефропатия.
IgA нефропатия (болезнь Берже) сейчас признается самой распространенной причиной хронической почечной недостаточности во всем мире. Болезнь Берже обнаруживается приблизительно у 10% больных с нефротическим синдромом. Наиболее часто она встречается у мужчин в возрасте от 10 до 30 лет. Ее признаками являются:
- поражение детей и подростков;
- эпизодическая гематурия, совпадающая по времени с респираторными инфекциями;
- легкая протеинурия, иногда нефротический синдром;
- гипертензия;
- повышение в сыворотке уровня IgA.
Этиология неизвестна, однако имеются сообщения, указывающие на географическую предрасположенность, причем наиболее часто болезнь встречается во Франции, Австралии и Сингапуре. Имеется слабая связь с HLA-DR4, но большее внимание в исследованиях направлено на обнаружение факторов, которые являются причиной повышения IgA; предположительно, ими могут быть вирусы или пищевые белки.
При световой микроскопии обнаруживается увеличение количества клеток в мезангиуме и увеличение количества мезангиального вещества. При прогрессировании заболевания развивается склероз. При иммунофлюоресцентных методах исследования обнаруживается накопление IgA в мезангиуме в виде сливающихся между собой отдельных гранул. Также часто обнаруживается С3. При электронной микроскопии определяются электронно-плотные отложения.
Клинически болезнь проявляется протеинурией, гематурией, которая обычно появляется при воспалительных заболеваниях верхних дыхательных путей.
Прогноз. При слабой фокальной мезангиальной пролиферации с накоплением IgA и С3 в мезангиуме всех клубочков прогноз хороший. С другой стороны, при развитии мезангиокапиллярного (мембранопролиферативного) типа поражения, иногда с сегментарным некрозом, прогноз значительно ухудшается, нарушение функции почек нарастает очень быстро. Хотя болезнь прогрессирует довольно медленно, отдаленный прогноз плохой. У большинства больных в течение 6 лет развивается хроническая почечная недостаточность.
Мембранозный гломерулонефрит (МГН) имеет определенную гистологию, но множество причин. Как предполагается, морфологические изменения отражают размер и скорость формирования иммунных комплексов.
Этиология. У 85% больных не удается определить причину заболевания и, в данном случае, говорят об идиопатическом мембранозном гломерулонефрите. Наиболее частыми причинами вторичного мембранозного гломерулонефрита являются:
- инфекционные – сифилис, малярия, гепатит В, шистосомиаз, лепра;
- лекарственные – пеницилламин, золото, ртуть, героин;
- опухоли – злокачественные лимфомы, лимфома Ходжкина, бронхогенный рак легкого;
- системные заболевания соединительной ткани (системная красная волчанка, прогрессирующий системный склероз;
- другими состояниями (серповидно-клеточная анемия).
Очень важно выявлять больных, у которых причиной заболевания почек являются выше перечисленные заболевания, т.к. после устранения причины почечные проявления утихают.
Во всех клубочках обнаруживается утолщение стенок капилляров без признаков пролиферации и воспаления. При иммунофлюоресценции обнаруживаются гранулярные отложения IgG и С3 в утолщенной капиллярной стенке. При электронной микроскопии определяется отложение электронно-плотных масс под эпителиальными клетками на внешней стороне базальной мембраны.
Различают три стадии заболевания (рис. 5):

Рис. 5. Мембранозный гломерулонефрит
Стадия I характеризуется субэпителиальным накоплением электронно-плотных куполообразных отложений (“горбы”). На этой стадии мембрана выглядит почти нормальной, что представляет трудности для дифференциального диагноза при световой микроскопии с гломерулонефритом с минимальными изменениями. Проникновение белков сквозь фильтрационную мембрану приводит к слиянию и исчезновению отростков подоцитов, но при мембранозном гломерулонефрите обязательно обнаруживается накопление электронно-плотных иммунных комплексов.
Стадия II характеризуется появлением остроконечных выростов на базальной мембране в сторону эпителия, проникающих между отложениями, которые заметно увеличиваются. Эти выступы можно обнаружить на световой микроскопии при окраске серебром (отложения при этом не окрашиваются).
На стадии III выросты увеличиваются и сливаются с эпителиальной стороны отложений; при окраске серебром базальная мембрана выглядит расщепленной, двухслойной, слои которой соединены выростами (это создает картину цепи, где неокрашенные зоны представляют собой отложения иммунных комплексов). На этой стадии при обычной световой микроскопии обнаруживается утолщение базальной мембраны.
При чисто мембранозном гломерулонефрите в клубочке не обнаруживается пролиферация каких-либо клеток. При прогрессировании заболевания в результате утолщения мембраны клубочек превращается в гиалиновую массу. Морфологические изменения при идиопатическом и вторичном мембранозном гломерулонефрите сходны. По мере прогрессирования заболевания стенка все более утолщается в результате накопления отложений, которые, в конце концов, подвергаются разрушению и лизису. В результате пораженный клубочек склерозируется.
Клинические проявления. МГН выявляется во всех возрастных группах, но все-таки чаще болеют взрослые (наиболее часто – в возрасте около 35 лет). Мужчины поражаются несколько чаще женщин. Клинически мембранозный гломерулонефрит может проявляться как нефротическим синдромом, так и асимптоматической протеинурией. Протеинурия является неселективной. Гематурия на ранних этапах обычно отсутствует. Циркулирующие иммунные комплексы обнаруживаются редко. Гипертензия наблюдается у половины больных. Лечение кортикостероидами в большинстве случаев не дает эффекта, почечная недостаточность в результате гломерулосклероза развивается в течение 2-20 лет. У большинства больных наблюдается медленная прогрессия с развитием хронический почечной недостаточности. По современным данным 70% больных живут более 10 лет. Прогноз лучше у женщин и еще лучше у детей. При вторичном МГН прогноз при излечении основного заболевания отличный. При МГН часто наблюдается повышение свертываемости крови, что может привести к тромбозу почечной вены.
Мембранопролиферативный (мезангиокапиллярный) гломерулонефрит ( МПГН) характеризуется наличием двух признаков: утолщения стенки капилляров и пролиферации мезангиальных клеток. При нем также подчеркивается дольчатая структура клубочка.
МПГН не является диагностическим маркером для одного заболевания; это только определение конкретного типа поражения почек при воздействии различных факторов. Он был впервые описан в 1965 году, при этом были выделены два основных типа (рис. 6); через 10 лет был описан редко встречающийся третий тип, но он не имеет особых клинических проявлений.

Рис.6. Мембранопролиферативный гломерулонефрит
Тип I мембранопролиферативного гломерулонефрита (с субэндотелиальными отложениям)
I тип МПГН обусловливается иммунокомплексным повреждением; он также называется мезангиокапиллярным гломерулонефритом (МКГН) с субэндотелиальными отложениями. Этот тип развивается при различных состояниях – инфекциях, опухолях, системных заболеваниях соединительной ткани, дефиците комплемента, реакциях на лекарства и генетических нарушениях. Однако у большинства больных не удается выяснить причину заболевания. Заболевание проявляется нефротическим синдромом, однако иногда развивается и гематурия. У 2/3 пациентов определяется гипокомплементемия. Хроническая почечная недостаточность развивается в течение 10 и более лет.
При световой микроскопии обнаруживается диффузное утолщение капиллярной стенки и пролиферация мезангиальных клеток. Базальная мембрана обычно расщеплена (с двойным контуром или в виде трамвайных рельсов). При иммунофлюоресценции обнаруживается отложение IgG и С3 в капиллярной стенке. При электронной микроскопии диагностическим является субэндотелиальное отложение иммунных комплексов.
Клинически I тип является болезнью детей и подростков, у которых развивается нефротический синдром или смешанный (нефротически-нефритический) синдром. В крови обычно снижается уровень С3. Скорость прогрессии варьирует, но в целом прогноз плохой.
Тип II мембранопролиферативного гломерулонефрита (“болезнь плотных отложений”).
Патогенез II типа МПГН, несмотря на то, что он еще не полностью раскрыт, связан с активацией комплемента по альтернативному пути без участия иммунных комплексов. Он часто развивается при различных инфекциях, однако отсутствие иммунных комплексов в электронноплотных отложениях исключает участие в развивающемся процессе иммунных комплексов. Поэтому иногда используется альтернативное название – болезнь плотных отложений.
При световой микроскопии обнаруживается эозинофильная, преломляющая свет, однообразно утолщенная базальная мембрана. Пролиферация мезангиальных клеток менее выражена, чем при I типе. При иммунофлюоресцентном исследовании обнаруживается отложение С3 в сосудистой стенке и мезангиуме. Иммуноглобулины не определяются.
Чаще поражаются дети и подростки. Клинические проявления идентичны таковым при I типе. Уровень С3 в сыворотке снижен, однако уровни С1q, С2 и С4 в пределах нормы, что является подтверждением того, что комплемент активируется по альтернативному пути. Практически у всех больных в крови обнаруживается С3 нефритогенный фактор. Механизм связи его с повреждением клубочков неизвестен. Прогноз плохой.
Патологическая анатомия конечной стадии хронического гломерулонефрита
Макроскопически почки значительно уменьшены в размерах, кора истончена и имеет неровную, зернистую поверхность (“вторично-сморщенная почка”). На разрезе кора неравномерно истончена, граница между корой и мозговым веществом нечеткая, сосуды почки хорошо выражены из-за утолщения их стенок.
Микроскопически в истонченной коре определяется значительное уменьшение количества нефронов, диффузный склероз клубочков, многие из которых превращаются в гиалинизированные шары. Лежащие между ними канальцы атрофируются, оставшиеся канальцы расширяются и заполняются розовым белковым материалом (“тироидизация”). Часто развивается выраженный интерстициальный фиброз.
При иммунофлюоресценции и электронной микроскопии изменения могут быть различными. В некоторых склерозированных клубочках обнаруживаются отложения электронно-плотных комплексов IgA, IgG и С3. Нахождение этих отложений важно для диагностики хронического гломерулонефрита, т.к. при хроническом пиелонефрите и гипертензионном нефросклерозе, которые макро- и микроскопически могут быть похожими, таких отложений никогда не бывает.
Клинически у больных с хроническим гломерулонефритом часто наблюдается гипертензия, микрогематурия, протеинурия и иногда нефротический синдром.
Осложнением гломерулонефрита при его остром и подостром течении может быть олиго- или анурия. Для хронического течения гломерулонефрита характерна хроническая почечная недостаточность с проявлениями азотемической уремии. Возможны также сердечно-сосудистая недостаточность, кровоизлияние в мозг, которые становятся причиной смерти.
Фокальный гломерулосклероз
Фокальный гломерулосклероз является важной патологией, так как он является причиной нефротического синдрома у 10% детей и 15% взрослых. Причина неизвестна. Патогенез неизвестен; некоторые авторы считают его осложнением заболевания с минимальными изменениями. У некоторых больных фокальный гломерулосклероз связан с внутривенным введением героина, несколько случаев заболевания было описано у ВИЧ-инфицированных больных, у которых вирус обнаруживали в эпителиальных клетках клубочков и канальцев.
Структурные изменения. Фокальный гломерулосклероз характеризуется присутствием фокальных сегментарных склеротических зон (розовый гиалиновый материал) в периферической части клубочка. В склеротических зонах часто обнаруживаются капельки жира. Первоначально поражаются клубочки в юкстамедуллярной зоне (глубоко в коре), поэтому при поверхностной биопсии почки они не попадают в срез, в результате чего данным больным ошибочно ставиться диагноз гломерулонефрита с минимальными изменениями (предполагается, что из-за данной ошибки при биопсии большинство случаев “стероидо-резистентного” гломерулонефрита с минимальными изменениями представляют собой именно фокальный гломерулосклероз). При иммунофлюоресцентных методах исследования в пораженных клубочках находят гранулярные отложения IgM, С3, иногда IgA и фибриногена. При электронной микроскопии определяется увеличение количества мезангиального матрикса и спадение капилляров клубочка в зонах гломерулосклероза. Отростки эпителиальных клеток капсулы Боумена сливаются между собой и исчезают. В некоторых местах эпителиальный покров исчезает. Предполагается, что ведущую роль в патогенезе играет повреждение эндотелиальных клеток.
Клинические проявления. При фокальном гломерулосклерозе проявления могут варьировать от асимптоматической протеинурии до нефротического синдрома. Протеинурия является неселективной. Прогноз плохой, болезнь медленно прогрессирует с развитием хронической почечной недостаточности в течение 10 лет. Лечение кортикостероидами неэффективно. У некоторых больных подобное заболевание развивается в пересаженной почке.
ВТОРИЧНЫЕ ПРИОБРЕТЕННЫЕ ГЛОМЕРУЛОПАТИИ
Гломерулопатии являются проявлением многих заболеваний соединительной ткани и васкулитов. Основные проявления этих заболеваний рассмотрены в отдельной лекции, а здесь суммированы почечные проявления этих заболеваний.
Вторичные гломерулонефриты могут быть обусловлены:
- иммунокомплексным повреждением;
- метаболическими нарушениями;
- поражением сосудов.
Иммунокомплексное повреждение почек
Иммунокомплексное повреждение может наблюдаться при следующих заболеваниях:
- системной красной волчанке;
- пурпуре Шенляйн-Геноха.
Системная красная волчанка (СКВ)
Почки при СКВ поражаются в 70% случаев. Волчаночный нефрит является наиболее частой причиной смерти при СКВ. Клинически он проявляется протеинурией, микрогематурией, нефротическим синдромом, острым нефритическим синдромом и почечной недостаточностью. В активную фазу заболевания уровень комплемента в сыворотке снижен.
При световой микроскопии могут обнаруживаться различные изменения (табл. 3). Наиболее выраженным изменением при световой микроскопии является фокальная или диффузная пролиферация эндотелиальных клеток капилляров клубочков. Часто наблюдается пролиферация мезангиальных клеток, но она не представляет особой опасности. В результате фокального или диффузного утолщения базальной мембраны возникает характерная картина при СКВ – образование “проволочных петель”. Иногда в зонах клубочковых изменений могут определяться специфические для СКВ базофильные (гематоксилиновые) тельца. При иммунофлюоресценции определяются гранулярные отложения IgG и С3 в базальной мембране клубочков и мезангиуме. При электронной микроскопии большие глыбки иммунных комплексов определяются в мезангиуме, под эндотелием и под эпителием. При наличии “проволочных петель” в клубочках определяются большие субэндотелиальные отложения иммунных комплексов, что позволяет отдифференцировать эту форму от мембранозного гломерулонефрита, при котором иммунные комплексы откладываются субэпителиально.
Таблица 3
Категории поражения почек при СКВ по определению ВОЗ
| Kласс | Патологические изменения | % | Kлинические проявления |
| I | Без изменений | Легкая степень поражения с микрогематурией и протеинурией; медленная прогрессия | |
| II | Мезангиальный гломерулонефрит | ||
| III | Фокальный пролиферативный гломерулонефрит | ||
| IV | Диффузный пролиферативный гломерулонефрит | Тяжелое поражение с быстрой прогрессией в ХПН | |
| V | Диффузный мембранозный гломерулонефрит | Нефротический синдром; медленная прогрессия в ХПН |
Клинические проявления и прогноз зависят от гистологического класса заболевания (табл. 3). Со временем у большинства больных с СКВ развивается хроническая почечная недостаточность.
Пурпура Шенляйн-Геноха
Пурпура Шенляйн-Геноха – это системный васкулит, поражающий кожу, суставы, кишечник и почки, развивается чаще всего он у детей. Кожная сыпь чаще всего располагается на разгибательных поверхностях конечностей и ягодицах. Также наблюдаются боли в суставах и кишечнике, иногда с кишечными кровотечениями.
Почки поражаются довольно часто, что проявляется гематурией, протеинурией, острой почечной недостаточностью и нефротическим синдромом. При световой микроскопии обнаруживается увеличение количества клеток в мезангиуме и формирование эпителиальных полулуний. При иммунофлюоресцентных методах исследования обнаруживается накопление IgA в мезангиуме. При электронной микроскопии определяются увеличение количества клеток в мезангиуме и электронно-плотные отложения.
Пурпура Шенляйн-Геноха является прогрессирующим заболеванием, у 20% больных развивается хроническая почечная недостаточность.
Метаболические нарушения
Метаболические нарушение, приводящие к поражению почек наблюдаются при:
- сахарном диабете;
- почечном амилоидозе;
Диабетическая нефропатия
При сахарном диабете происходит повреждение как крупных, так и мелких сосудов во всем организме. В результате вовлечения в процесс сосудов микроциркуляторного русла поражаются сетчатка глаза, нервы, кожа и особенно почки, что приводит к развитию гломерулопатии, гиалинозу артериол и поражению канальцевого аппарата. Комбинация этих поражений может быть различной, но все они объединяются в понятие диабетической нефропатии.
По мере прогрессии основного заболевания усиливается протеинурия, что может привести к нефротическому синдрому и почечной недостаточности. Приблизительно 10% больных сахарным диабетом умирают от почечной недостаточности. Однако, если диабет развивается в детском возрасте (обычно инсулин-зависимый тип), то смерть в результате почечной недостаточности наблюдается у 50% больных.
Патогенез. Все изменения развиваются в базальной мембране, проницаемость которой резко увеличивается. Увеличение проницаемости может происходит в результате:
- избытка коллагена IV типа в базальной мембране;
- дефицита протеогликанов, таких как гепарансульфата, которые обеспечивают полианионную природу мембраны;
- неферментного гликозилирования белков; это происходит в результате гипергликемии, что может нарушать полианионную структуру базальной мембраны и также нарушать физико-химические характеристики циркулирующих белков;
- гиперфильтрации в результате увеличенного почечного кровотока, обнаруживаемого у диабетиков.
Макроскопически почки обычно мало изменены, однако в тяжелых случаях они могут быть сморщенными и иметь мелкобугристую поверхность.
Гистологически могут обнаруживаться три типа изменений, которые отражают степень тяжести поражения:
- утолщение стенки капилляров на начальных этапах;
- увеличение объема мезангиального матрикса, что приводит к сдавлению сосудов клубочка и развитию диффузного гломерулосклероза;
- для диабета очень характерно узловое увеличение объема мезангиума среди долек клубочка и обозначается как узелковый гломерулосклероз или изменения Киммельстила-Вильсона.
Поражение клубочков сочетается с гиалинозом сосудом, поражающем как приносящие, так и выносящие артериолы. При электронной микроскопии при фокальном узловом типе поражения обнаруживается увеличение количества мезангиального матрикса. Депозиты отсутствуют, поэтому при иммунофлюоресценции никаких изменений не находят.
У больных диабетом часто развивается острый пиелонефрит. У таких больных часто встречается папиллярный некроз. Папиллярный некроз (инфаркт сосочков) обусловлен ишемией. Обтурационная ишемия связана с утолщением стенок тонких прямых сосудов в результате их диабетического повреждения в сочетании с проявлениями воспаления. Некротизированные сосочки могут отрываться и выходить с мочой или вызывать обструкцию мочевыводящих путей.
Заболевание быстро прогрессирует, приводя к развитию хронической почечной недостаточности.
Амилоидоз почек
Почки поражаются в 80-90% случаев вторичного амилоидоза и в приблизительно 30% при первичном амилоидозе. У таких больных обнаруживается выраженная протеинурия или нефротический синдром. Почечные проявления у 50% больных являются первым проявлением заболевания.
В почках амилоид откладывается в стенках сосудов, в капиллярных петлях, что сопровождается утолщением базальной мембраны, в мезангиуме клубочков, в базальных мембранах канальцев и в строме. При электронной микроскопии выявляется специфический фибриллярный белок амилоида (АА-амилоид). Почки становятся плотными, большими и “сальными”. По мере нарастания процесса клубочки и пирамиды полностью замещаются амилоидом, разрастается соединительная ткань и развивается амилоидное сморщивание почек.
Патологическая анатомия. В течении амилоидоза почек различают следующие стадии:
- латентную;
- протеинурическую;
- нефротическую;
- азотемическую (уремическую).
В разные стадии изменения почек различны и отражают динамику процесса.
В латентной стадии внешне почки изменены мало, хотя в пирамидах (сосочках) обнаруживаются склероз и отложение амилоида по ходу прямых сосудов и собирательных трубок. Изменения клубочков состоят в утолщении и двухконтурности мембран их капилляров, просветы которых аневризматически расширены. В интермедиарной зоне и пирамидах строма пропитана белками плазмы.
В протеинурической стадии амилоид появляется не только в пирамидах, но и в клубочках в виде небольших отложений в мезангиуме и отдельных капиллярных петлях, а также в артериолах. Склероз и амилоидоз пирамид и пограничного слоя резко выражены и ведут к атрофии многих глубоко расположенных нефронов, редукции путей юкстамедуллярного кровотока и лимфотока в мозговом веществе почек. Эпителий канальцев главных отделов в состоянии гиалиново-капельной или гидропической дистрофии; в просвете канальцев обнаруживаются цилиндры. Почки увеличены, плотны, поверхность их бледно-серая или желто-серая. На разрезе корковый слой широкий, матовый, мозговое вещество серо-розовое, “сального” вида, нередко цианотичное (большая сальная почка).
В нефротической стадии количество амилоида в почках увеличивается. Он обнаруживается во многих капиллярных петлях большинства клубочков, в артериолах и артериях, по ходу собственной мембраны канальцев, однако выраженный склероз коркового вещества отсутствует. Канальцы расширены, заполнены цилиндрами. В эпителии канальцев в строме много липидов (холестерина). Почки имеют вид, типичный для так называемого амилоидно-липоидного нефроза. Они становятся большими, плотными, восковидными – большая белая амилоидная почка.
В азотемической (уремической) стадии в связи с нарастающим амилоидозом и склерозом наблюдаются гибель большинства нефронов, их атрофия, замещение соединительной тканью. Почки обычных размеров или несколько уменьшены. Они очень плотные, со множеством рубцовых западений на поверхности (амилоидно-сморщенные почки) В этой стадии нередко развивается гипертрофия сердца, особенно левого желудочка, что связано с развитием нефрогенной артериальной гипертензии.
Клинически, отложение амилоида проявляется повышением сосудистой проницаемости гломерулярных капилляров, что приводит к развитию нефротического синдрома.
Амилоидоз довольно быстро прогрессирует. Хроническая почечная недостаточность является основной причиной смерти больных амилоидозом.
Сосудистое повреждение
Повреждение клубочков почек наблюдается при следующих сосудистых заболеваниях:
- узелковом периартериите;
- гранулематозе Вегенера;
- гемолитико-уремическом синдроме;
- идиопатической тромбоцитопенической пурпуре;
- синдроме диссеминированного внутрисосудистого свертывания (ДВС-синдроме).
Характер поражения при системных васкулитах один и тот же – сегментарный некроз, иногда с формированием полулуний. У таких больных часто обнаруживаются аутоантитела против цитоплазматических антигенов нейтрофилов (ANCA), которые могут быть двух классов: цитоплазматические (С-) ANCA, которые реагируют с протеиназой 3, и перинуклеарные (Р–) ANCA, которые реагируют с миелопероксидазой. У большинства больных с гранулематозом Вегенера обнаруживаются С-ANCA, а у больных с узелковым периартериитом – Р-ANCA, однако в последнем случае они являются менее специфическими. При тромботических микроангиопатиях в капиллярах клубочков обнаруживаются фибрин или тромбоциты, или и то, и другое.
Узелковый периартериит. Поражение почек встречается у 80% больных с узелковым периартериитом. 30% больных умирают в результате ХПН.
Макроскопически почки уменьшены в объеме, на их поверхности имеются признаки инфарктов и множественных кровоизлияний. При световой микроскопии в стенках сегментарных и дугообразных артерий почек определяются фибриноидный некроз, воспаление, тромбоз, образование аневризм и их разрыв. При хроническом течении в сосудах наблюдается фиброз, который является неспецифичным для заболевания.
Микроскопически в клубочках обнаруживаются участки фибриноидного некроза и образование эпителиальных полулуний. В зонах фибриноидного некроза при иммунофлюоресценции находят фибрин и иммуноглобулины (в основном IgG). Т.к. у многих больных узелковым периартериитом обнаруживается в сыворотке поверхностный антиген вируса гепатита В, то предполагается, что в развитии поражений почек у некоторых больных принимают участие иммунные комплексы, образующиеся при взаимодействии антител с этим антигеном.
Клинически поражение почек проявляется гематурией, протеинурией и гипертензией. При отсутствии лечения больные умирают в сроки от месяца до нескольких месяцев или лет, но при проведении иммуносупрессивной терапии прогноз значительно улучшается. Почечная недостаточность развивается обычно довольно быстро.
Гранулематоз Вегенера может встречаться в любом возрасте, однако наиболее часто – в 40-60 лет, при этом чаще у мужчин. Поражение почек является одной из частей классической триады признаков гранулематоза Вегенера – двумя другими является поражение верхних дыхательных путей и легких. Клинические проявления варьируют от микроскопической гематурии до быстро прогрессирующей почечной недостаточности. Антинейтрофильные цитоплазматические антитела обнаруживаются у 95% больных. Поражение почек развивается у 90% больных и проявляется протеинурией и гематурией, при этом наблюдается довольно быстрое прогрессирование в почечную недостаточность.
При световой микроскопии определяется некротизирующий гранулематозный артериит, поражающий артерии малого и среднего калибра. В клубочках обнаруживается фокальный сегментарный пролиферативный гломерулонефрит. Часто также обнаруживается фибриноидный некроз, тромбоз капилляров и образование эпителиальных полулуний. При иммунофлюоресценции определяется накопление IgA, С3 и фибриногена в стенке капилляров клубочка.
При лечении циклофосфамидом удается вызвать ремиссию у большинства больных, при этом при раннем лечении возможно полное восстановление функции почек.
Гемолитико-уремический синдром (ГУС) – это сложное состояние, которое характеризуется:
- острой нефропатией;
- гемолизом;
- тромбоцитопенией.
Волокна фибрина накапливаются в мелких сосудах, в том числе и капиллярах клубочков. В образующейся сети, через которую продолжает протекать кровь, деформируются эритроциты и тромбоциты, что приводит к их деструкции. Этот процесс назван микроангиопатическим гемолизом. Выделяют три типа ГУС: детский, взрослый и вторичный.
Детский тип ГУС. При детском типе ГУС прогноз намного лучше, чем у взрослых. Часто предшествует продромальный период, который характеризуется диареей или ОРВИ в течение 5-15 дней. Затем внезапно развивается олигурия, гематурия и иногда мелена. В крови определяется анемия. Приблизительно у половины больных развивается гипертензия. Патогенез точно не выяснен, но географическая локализация заболевания говорит о наличии специфического инфекционного агента; в некоторых случаях доказано роль Escherichia coli, продуцирующей веротоксин.
Взрослый тип ГУС. У взрослых ГУС обычно является летальным и может развиться при следующих ситуациях:
- беременности – иногда развивается в послеродовом периоде, даже через несколько месяцев после родов;
- терапии эстрогенами – наблюдается у женщин, использующих оральные контрацептивы и, реже, у мужчин, принимающих эстрогены при лечении рака предстательной железы;
- инфекциях, таких как тиф, некоторых вирусных инфекциях и шигеллезах.
Вторичный ГУС наблюдается как осложнение:
- злокачественной гипертензии;
- прогрессирующего системного склероза;
- красной системной волчанки;
- отторжения трансплантата.
Клинически и морфологически вторичный ГУС идентичен с другими типами.
Гистологические характеристики ГУС. В капиллярах клубочков обнаруживаются тромбы, отмечается сегментарное набухание эндотелиальных и мезангиальных клеток. Стенка капилляров утолщена. В артериолах и мелких артериях обнаруживается фибрин и эритроциты, часто они тромбируются. При значительном тромбозе сосудов развиваются инфаркты коры почек.
Идиопатическая тромбоцитопеническая пурпура (ИТП) развивается чаще у женщин, в основном после 40 лет. Проявляется неврологическими симптомами, которые сочетаются с гемолитической анемией и тромбоцитопенией. Поражение почек развивается у половины больных и проявляется протеинурией, микрогематурией и нарушением функции почек.
Микроскопически в капиллярах клубочков, афферентных артериолах и междольковых артериях определяются эозинофильные гранулярные белые тромбы.
Синдром диссеминированного внутрисосудистого свертывания ДВС-синдром характеризуется генерализованным повреждением эндотелия сосудов. Наиболее частой причиной является грам-отрицательная септицемия. Фибриновые тромбы заполняют просвет капилляров клубочков, афферентных артериол и мелких артерий, что является причиной тяжелого почечного поражения.
ТУБУЛОПАТИИ
Тубулопатии представляют собой заболевания почек с первичным ведущим поражением канальцев, которое сопровождается нарушением их концентрационной, реабсорбционной и секреторной функции. Различают приобретенные и наследственные тубулопатии (обусловленные различными формами ферментопатий). К приобретенным тубулопатиям относят: острая почечная недостаточность (ОПН) и хронические тубулопатии (миеломная и подагрическая почка).
Морфологическим субстратом ОПН является острый некротический нефроз (ОНН); у больных при этом развивается резкая олигурия (менее 100 мл мочи в сутки). Основными причинами ОПН являются ишемия и токсическое поражение.
Ишемический ОНН может развиться при различных состояниях, например, травмах, ожогах, инфекциях, при которых развивается шоковое состояние и коллапс. Выраженная гипотензия является причиной критического понижения кровотока в почках, особенно в кортикальных клубочках в связи с особенностями их кровоснабжения. Почки выглядят бледными и отечными. При гистологическом исследовании определяется повреждение эпителиальных клеток канальцев практически по всей их длине, часто развивается вакуольная дистрофия, разрыв канальцевой базальной мембраны (тубулорексис). Интерстициум становиться отечным. Часто образуются цилиндры в дистальных канальцах и собирательных трубочках; цилиндры состоят из клеточного детрита и белков. При краш-синдроме в цилиндрах обнаруживается миоглобин, а при нарушении правил переливания крови – гемоглобин.
Токсический ОНН может развиться при действии следующих веществ:
- тяжелых металлов (свинца, ртути, мышьяка, золота, хрома, висмута и урана);
- органических растворителей (четыреххлористого углерода, хлороформа);
- гликолей (этиленгликоля, пропиленгликоля и др.);
- лекарственных веществ (антибиотиков – метациклина, сульфаниламидов, полимиксина, цефалоспоринов; нестероидных противовоспалительных препаратов; ртутных диуретиков и др.);
- йодсодержащих рентгенконтрастных веществ;
- фенола;
- пестицидов;
- бактериальных токсинов.
Почки выглядят набухшими и красными. Гистологически, в эпителиальных клетках канальцев развивается вакуольная дистрофия и некроз, наиболее выраженные в проксимальных канальцах, дистальные канальцы поражаются в меньшей степени. Этим отличается поражение при токсическом ОНН от ишемического ОНН, при котором поражаются канальцы по всей длине.
Различают две стадии развития заболевания. После стадии анурии, которая развивается из-за перекрытия просвета канальцев набухшими и некротизированными клетками, наступает полиурическая стадия, связанная с нарушением концентрационной способности почек. В олигурическую стадию большой риск для жизни больного представляет развивающаяся гиперкалиемия, которая может привести к аритмии сердца. Однако в начале полиурической стадии может развиться, наоборот, гипокалиемия.
Исход. Если состояние будет вовремя диагностировано и будет проведена адекватная инфузионная терапия и гемодиализ больной может полностью выздороветь. Эпителий канальцев регенерирует, если сохранена базальная мембрана. При неблагоприятном развитии процесса больные умирают от острой почечной недостаточности.
ТУБУЛО-ИНТЕРСТИЦИАЛЬНЫЕ ЗАБОЛЕВАНИЯ ПОЧЕК
Тубуло-интерстициальные заболевания – это заболевания почек, которые характеризуются первичным поражением интерстициума и почечных канальцев.
Термин “интерстициальный нефрит” используется для обозначения группы состояний, имеющих сходные клинические и морфологические характеристики, но разные причины. Морфологические проявления характеризуются воспалительной реакцией, при которой в интерстициальной ткани преобладают Т-клетки. Патогенез во многих случаях не выяснен. Поэтому классификация основана на этиологических факторах:
- токсический – в результате действия солей тяжелых металлов (свинца, ртути, золота и др.) и некоторых лекарств (гентамицина, цефалоридина, циклоспорина А);
- инфекционный (вирусы, бактерии);
- иммунологический;
- метаболический – ураты;
- механический – обструкция;
- неопластический – миелома.
Проявлением острого интерстициального нефрита является острая почечная недостаточность; для определения причины необходимо тщательно собрать анамнез о приеме различных веществ, которые могут привести к поражению почек. Некоторые вещества могут быть причиной как острого некроза канальцев, так и острого интерстициального нефрита.
Гистологически определяется отечность интерстициальной ткани, инфильтрация мононуклеарами (лимфоцитами, макрофагами, плазматическими клетками) и дистрофия канальцев различной степени выраженности.
Больные с хроническим интерстициальным нефритом обычно обращаются к врачу уже с развившейся хронической почечной недостаточностью. Гистологически определяется выраженный интерстициальный фиброз, атрофия канальцев и лимфогистиоцитарная инфильтрация.
Анальгетическая нефропатия является хорошо изученной неблагоприятной реакцией на анальгетики. Впервые большая вспышка заболевания была описана в 1953 году в Швейцарии. При эпидемиологических и экспериментальных исследованиях было установлено, что употребление в больших дозах аспирина в сочетании с фенацетином является потенциально опасным. Из фенацетина образуются метаболиты, которые способны связываться с клеточными белками, что приводит к истощению запасов глютатиона в клетках. Аспирин вызывает ишемию сосочков почек, т.к. он ингибирует синтез простагландинов, обладающих вазодилатационным эффектом. Токсический и ишемический эффект, таким образом, суммируется, что приводит к селективному повреждению вплоть до некроза сосочков почек.
Также установлено, что у пациентов с анальгетической нефропатией чаще развивается переходноклеточный рак почечной лоханки и мочеточников.
При различных метаболических расстройствах может наблюдаться поражение канальцев и интерстициума. Наиболее часто встречаются:
- гипокалиемическая нефропатия;
- уратная нефропатия;
- гиперкальциемическая нефропатия;
- оксалатная нефропатия.
Гипокалиемическая нефропатия. Выраженная гипокалиемия может развиться при:
- хронической диарее;
- гиперальдостеронизме, как первичном, так и вторичном;
- длительном применении слабительных и диуретиков.
Гипокалиемия приводит к тяжелой гидропической дистрофии клеток проксимальных канальцев, прямые канальцы и собирательные трубочки не поражаются.
Уратная (подагрическая) нефропатия. Поражение почек при подагре происходит из-за высокого содержания мочевой кислоты в крови. Кроме того, у больных с хронической почечной недостаточностью в результате пиелонефрита или гломерулонефрита нарушается фильтрация и канальцевая секреция мочевой кислоты. Это приводит к накоплению мочевой кислоты в крови и отложению ее в почках. Мочевая кислота кристаллизуется в кислой среде, которая наблюдается в дистальных канальцах, собирательных трубочках и интерстициуме сосочков.
Острая уратная нефропатия проявляется в виде острой почечной недостаточности в основном у больных с миелопролиферативными заболеваниями. Она часто провоцируется химиотерапией, после проведения которой происходит гибель значительного количества клеток, при этом в кровь высвобождаются огромные количества нуклеиновых кислот. На поверхности среза пораженных почек обнаруживаются желтые полосы в мозговом слое почек в результате отложения кристаллов уратов, которые заполняют просвет канальцев; в результате чего происходит обструкция и дилатация вышележащих канальцев.
Хроническая уратная нефропатия встречается намного чаще и наблюдается у больных с повышенным содержанием мочевой кислоты в крови, например при подагре. Кристаллы в просвете канальцев являются причиной хронической обструкции и тубуло-интерстициального нефрита в коре, которая становиться атрофичной и истонченной.
Уратные камни могут образовываться и при острой, и при хронической уратной нефропатии, что приводит к увеличению заболеваемости пиелонефритом.
Гиперкальциемическая нефропатия (нефрокальциноз). При повышении уровня кальция в плазме наблюдается отложение кальция в почках – метастатическая кальцификация. Проявления являются необычными; нарушение функции канальцев приводит к нарушению концентрации мочи, что проявляется полиурией, часто сочетающейся с никтурией. На поверхности среза определяются камни в чашечно-лоханочной системе и линейные белые полосы и пятна. При микроскопии определяется интерстициальный фиброз, неспецифическая воспалительная инфильтрация и атрофия канальцев в сочетании с кальцификацией.
Оксалатная нефропатия. При повышении содержания в крови оксалата кальция происходит отложение его в тканях. В почках наблюдается усиление экскреции оксалатов, что приводит к повреждению почек. Гипероксалатемия и, соответственно, гипероксалатурия могут быть первичными и вторичными. Первичные формы встречаются при дефиците ферментов печени, которые участвуют в декарбоксилировании глиоксалата, которые накапливается и окисляется с образованием оксалатов. Вторичная гипероксалатурия развивается при отравлении этиленгликолем (антифризом) или некоторыми анестетиками (метоксифлюраном) и дефиците пиридоксина (витамина В6). Отложение оксалатов в почках наблюдается также при некоторых хронических заболеваниях почек.
При тяжелых случаях почки уменьшены в размере, имеют бугристую поверхность и сморщены, кора истончена в результате фиброза и разрушения канальцев, которые развиваются в результате отложения оксалатов. В чашечках и лоханке часто обнаруживаются камни.
Поражения, связанные с действием физических агентов
Радиационный нефрит. Клетки почечных канальцев имеют высокую чувствительность к ионизирующему излучению, поэтому необходимо быть осторожными при терапевтическом облучении верхних отделов живота. У таких больных развивается гипертензия и почечная недостаточность.
Гистологически определяется гломерулосклероз, выраженное поражение сосудов и интерстициальный фиброз. Канальцы выстланы атипичными клетками и обнаруживается характерная для данного состояния многослойная базальная мембрана.
Обструктивная уропатия (гидронефроз) является частой причиной интерстициального нефрита. Причинами обструкции мочевыводящих путей могут быть:
- врожденные аномалии (мочеточниково-лоханочный стеноз, пузырно-мочеточниковый рефлюкс);
- опухоли (рак мочевого пузыря и предстательной железы);
- гиперпластические состояния (доброкачественная гиперплазия предстательной железы);
- камни.
Клинические симптомы и прогноз зависят от уровня обструкции. Так, например, камень или фрагмент оторвавшегося сосочка может стать причиной почечной колики, а рак мочевого пузыря или доброкачественная гиперплазия предстательной железы проявиться пузырными симптомами. Острая обструкция нижних отделов мочевого тракта приведут к анурии и болям.
Частичная или односторонняя обструкция может протекать скрытно, иногда бессимптомно в течении нескольких лет. Проявляется она полиурией и никтурией. На ранних стадиях определяется снижение концентрационной функции почек, канальцевый ацидоз и отложение большого количества солей. Часто развивается гипертензия.
Патогенез. Лоханка и чашечки становятся расширенными в результате повышения давления; при острой обструкции расширение выражено не сильно. Перистальтическая активность мочеточников повышается, что приводит к повышению внутрилоханочного давления, которое передается на паренхиму. Первоначально образующийся ультрафильтрат реадсорбируется через лимфатические и кровеносные сосуды. При продолжающемся быстром повышении давления происходит снижение кровотока в клубочках и мозговом слое, что приводит к нарушению фильтрации. В результате длительной обструкции развивается сильное расширение лоханки с истончением паренхимы (гидронефроз).
ПИЕЛОНЕФРИТ
Пиелонефрит – это инфекционное заболевание, при котором инфекция может попадать в почки гематогенным (нисходящим) или уриногенным (восходящим) путем. В результате обильного кровоснабжения почек они часто поражаются при различных состояниях, сопровождаемых септицемией. Наиболее часто возбудителями являются бактерии. Инфекции мочевых путей встречаются достаточно часто, занимая второе место после инфекций дыхательных путей. Однако не при всех инфекциях мочевыводящих путей может развиться пиелонефрит, для его развития необходим пузырно-мочеточниковый рефлюкс.
|
|
|
|
|
Дата добавления: 2014-01-11; Просмотров: 586; Нарушение авторских прав?; Мы поможем в написании вашей работы!