КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Физико-химические методы анализа веществ
Учебные вопросы:
1) Классификация и особенности физико-химических методов анализа веществ.
2) Оптические методы анализа веществ: фотометрия, спектральный анализ.
3) Электрохимические методы анализа.
4) Хроматографические методы анализа веществ.
Физико-химические методы анализа веществ основаны на наблюдении за изменением физических свойств анализируемого вещества, которое происходит в результате определенной химической реакции.
Специфическими особенностями физико-химических методов анализа веществ являются: использование эталона – стандартного вещества с известным содержанием анализируемого элемента, относительно которого определяется количество этого элемента в анализируемой пробе, а также применение измерительного прибора для регистрации аналитического сигнала.
В зависимости от наблюдаемого аналитического сигнала различают оптические, электрохимические и хроматографические методы анализа.
Оптические методы анализа изучают взаимодействие с веществом различных видов излучения. Как известно, вещества могут поглощать свет в видимой области (380 – 760 нм) и иметь окраску, отражать, пропускать и рассеивать излучение. В зависимости от поведения вещества при взаимодействии с излучением различают следующие оптические методы анализа:
1) фотометрию (включающую фотоколориметрию и спектрофотометрию);
2) турбидиметрию;
3) нефелометрию;
4) рефрактометрию.
Метод фотометрии основан на измерении интенсивности поглощенного веществом света в ультрафиолетовой (УФ), видимой и инфракрасной (ИК) областях спектра. Фотоколориметрия анализирует содержание (концентрацию) вещества на основе измерения поглощения им излучения видимой части спектра. Окраску исследуемого раствора сравнивают с окраской эталона (раствора известной концентрации). Измерения проводят с помощью прибора − фотоэлектроколориметра. Излучение видимой области спектра поглощают только окрашенные вещества, поэтому метод применим только для окрашенных растворов. Если изучаемое вещество не окрашено, предварительно его переводят в окрашенное соединение путем проведения химической реакции с определенными реагентами.
Спектрофотометрия анализирует вещества на основе измерения поглощения ими монохроматического излучения в УФ, видимой и ИК областях спектра. Измерения проводят с помощью прибора – спектрофотометра.
Теория метода фотометрии основана на законе светопоглощения Бугера – Ламберта – Бера, утверждающем, что при прохождении монохроматического светового потока через поглощающий его раствор интенсивность прошедшего света I отличается от интенсивности падающего света Iо. Отношение I: Iо называется пропусканием (прозрачностью) раствора. Величина десятичного логарифма этого отношения lg (I/Iо) является важной характеристикой поглощающего свет раствора, которую называют оптической плотностью А:
lg (I / Iо) = А
Уменьшение интенсивности проходящего света (оптическая плотность раствора) пропорциональна произведению концентрации вещества в растворе (с) на толщину поглощающего слоя раствора (b) и определяется из формулы:
А = ε∙b∙c (27),
где ε – молярный коэффициент поглощения раствора при данной длине волны. Как видно из формулы (1), при постоянной толщине поглощающего слоя b наблюдается прямо пропорциональная зависимость А от концентрации с.
По физическому смыслу ε – оптическая плотность раствора при концентрации поглощающего вещества 1 моль/л и толщине поглощающего слоя 1 см. Так как поглощение при разных длинах волн (λ) различно, молярный коэффициент поглощения вещества ε зависит от длины волны падающего света. При данной длине волны ε зависит от природы вещества, от температуры и не зависит от концентрации и толщины слоя раствора, т.е. при определенной длине волны и температуре молярный коэффициент поглощения представляет постоянную для данного соединения величину и является индивидуальной характеристикой вещества.
Для многих окрашенных соединений ε = 104. Минимальная оптическая плотность (Амин.), которая может быть измерена оптическим прибором, составляет 0,01. При толщине слоя раствора в 1 см минимальная определяемая концентрация вещества в растворе (смин.) определяется из уравнения (27) и составляет 10-6 моль/л.
Зависимость светопоглощения (А, ε или их логарифмов) от длины волны падающего светового потока носит название спектра поглощения вещества. Это важная индивидуальная характеристика вещества. Излучение одних длин волн поглощается в большей степени, чем других, в зависимости от оптических свойств молекул и ионов данного вещества. Спектр поглощения имеет определенное число полос поглощения. Каждая полоса характеризуется максимумом поглощения (А макс. или εмакс.) и шириной полосы (а), как показано на рис. 45.
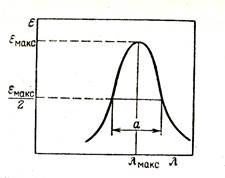
Рис. 45. Параметры полосы поглощения
Спектры поглощения одного и того же вещества в координатах А – λ имеют одинаковую форму независимо от толщины слоя и концентрации раствора и характеризуются максимумом при одной той же длине волны λ (см. рис. 46).
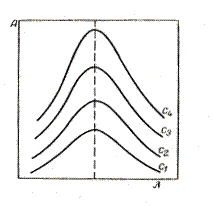
Рис. 46. Зависимость оптической плотности от длины волны при различных концентрациях вещества в растворе (с1 >с2 >с3 >с4)
Для измерения светопоглощения исследуемого раствора выбирают длину волны светового потока, соответствующую максимуму полосы поглощения (это обеспечивает наибольшую чуствительность и точность определения). Для этого измеряют оптическую плотность раствора при разных длинах волн и строят график в координатах А – λ. В спектрофотометрах зависимость оптической плотности А от длины волны λ записывается автоматически. В фотоэлектроколориметрах для получения монохроматического света применяют светофильтры – специальные стекла, пропускающие излучение определенного интервала длин волн (20 – 60 нм).
В соответствии с уравнением (27) увеличение толщины поглощающего слоя позволяет понизить предел обнаружения вещества в растворе. Но при толщине больше 5 см увеличиваются потери на рассеивание света. Поэтому для фотометрирования используют кюветы с толщиной слоя 1-5 см.
Если исследуемый компонент переводят в окрашенное соединение, измеряют светопоглощение последнего, причем условия проведения фотометрической реакции должны обеспечивать полноту образования и устойчивость полученного окрашенного продукта. Для выбора оптимальных условий изучают влияние рН на интенсивность окраски раствора при постоянных концентрациях исследуемого вещества и реагента. Небольшие изменения выбранного значения рН при фотометрировании (на рис. 47 пунктир) не должны влиять на светопоглощение. Постоянное значение рН поддерживают, как правило, при помощи буферных растворов.
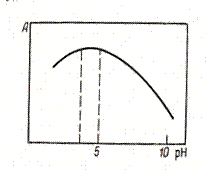
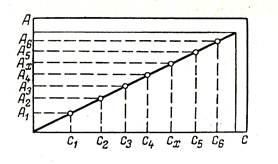
Рис. 47. Зависимость оптической Рис. 48. Градуировочный график
плотности от рН среды
Основным условием для количественного фотометрического определения является подчинение полученного при реакции окрашенного соединения закону Бугера – Ламберта – Бера. Для проверки выше сказанного в данном диапазоне концентраций строят градуировочный график зависимости оптической плотности А от концентрации с при выбранной ранее длине волны λ. Если закон светопоглощения соблюдается, то график представляет собой прямую линию, проходящую через начало координат (рис. 48). Для построения градуировочного графика готовят серию стандартных растворов определяемого вещества разной концентрации. При этом используют химически чистые вещества и дистиллированную воду. При выбранной длине волны и толщине слоя раствора измеряют оптические плотности стандартных растворов, которые наносят на градуировочный график. Измерив оптическую плотность анализируемого раствора Ах, по этому графику находят его концентрацию сх.
Спектрофотометрический метод дает возможность определить сразу несколько светопоглощающих веществ в одном растворе без предварительного разделения. Измеряя оптическую плотность этого раствора (Асмеси) при двух длинах волн λ1 и λ2 и используя правило аддитивности светопоглощения (каждое вещество делает свой вклад в экспериментальную величину оптической плотности раствора, т.е. Асмеси = А 1 + А 2 + …), путем решения системы уравнений:
Асмеси (для λ1) = ε1∙b∙c1 + ε2∙b∙c2
Асмеси (для λ2) = ε1∙b∙c1 + ε2∙b∙c2
можно рассчитать молярные концентрации веществ в анализируемом растворе.
В основе фотометрического метода анализа лежит измерение ослабления светового потока после прохождения его через раствор поглощающего вещества. Для этого сравнивают два потока: один, проходящий через исследуемый раствор, а другой – через раствор сравнения (содержащий все реактивы кроме определяемого вещества) или растворитель. Поглощение раствора сравнения принимают за нуль, а поглощение анализируемого раствора измеряют относительно раствора сравнения. Интенсивности световых потоков измеряют после преобразования в электрический сигнал с помощью фотоэлемента.
Количественный анализ по светопоглощению используется для определения содержания меди в аммиакате, ионов железа Fe2+ в растворе, марганца и хрома в их смесях.
Измерение светопоглощения в мутных средах (суспензиях и эмульсиях) лежит в основе метода турбидиметрии, использующего для определения химическую реакцию осаждения, например, сульфата бария:
Ba2+ + SO42- = BaSO4
Точность метода ниже, чем метода фотометрии, так как получение дисперсной системы с одинаковыми по размеру частицами представляет определенную трудность (может произойти коагуляция частиц). Ослабление интенсивности света, прошедшего через такие системы, также подчиняется основному закону светопоглощения.
На определении малых концентраций (не более 100 мг/л) вещества путем измерения интенсивности света, рассеянного частицами суспензии или эмульсии, основана нефелометрия. Этот метод применяют для анализа веществ (сульфата бария, хлорида серебра и др.), нерастворимых в воде, но образующих стойкие суспензии, для стабилизации которых необходимо добавлять защитный коллоид – раствор крахмала или желатина.
Интенсивность рассеянного света измеряется в направлении перпендикулярном падающему световому потоку с помощью прибора – нефелометра. Принцип его действия основан на уравнивании двух световых потоков, первого – от рассеивающего свет раствора, а второго – от стеклянного рассеивателя прибора.
В этом методе также используются стандартные растворы для приготовления суспензий, интенсивность светорассеяния которых сравнивается с интенсивностью светорассеивания исследуемых систем, быстро возрастающей с уменьшением длины волны падающего света. Интенсивность рассеянного света также зависит от размера частиц дисперсной системы, поэтому приготовление суспензии должно проводится в строго определенных условиях: с учетом концентрации, температуры, порядка и скорости смешения растворов и введения защитных коллоидов. Концентрацию анализируемого вещества находят по градуировочному графику, построенному для серии стандартных суспензий после их нефелометрирования.
Рефрактометрия применяется для определения содержания вещества или его состава путем измерения показателя преломления света.
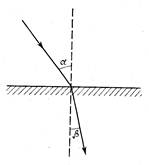 Рефракцией или преломлением называется изменение направления прямолинейного распространения луча света при переходе из одной среды в другую (см. рис. 49).
Рефракцией или преломлением называется изменение направления прямолинейного распространения луча света при переходе из одной среды в другую (см. рис. 49).
Рис. 49. Преломление светового
луча на границе раздела двух сред
Согласно закону преломления отношение синусов углов падения и преломления – величина постоянная для данного вещества, называемая показателем преломления (n): n = sin α / sin β.
Показатель преломления вещества зависит также от температуры (при повышении температуры уменьшается) и длины волны падающего света, с увеличением которой он уменьшается. Чаще всего его измеряют по отношению к воздуху (при падении света на преломляющую среду из воздуха). Рефрактометрические измерения проводят при 20оС и применяют монохроматический свет (с определенной длиной волны). Приборы, служащие для измерения n, называются рефрактометрами. Принципиальная схема преломления лучей в призмах прибора представлена на рис. 50.
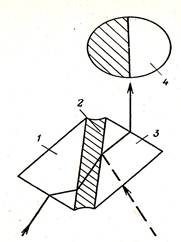
Рис. 50. Устройство рефрактометра:
1- осветительная призма;
2 - слой раствора;
3 – измерительная призма;
4 - поле зрения в зрительной трубе
В электрохимических методах анализа аналитический сигнал связан с изменением электрических и магнитных свойств анализируемых систем в зависимости от концентрации веществ. К таким свойствам относятся: количество электричества; электродный потенциал; электропроводность и другие. Измерение изменения таких свойств осуществляется с помощью внешнего источника постоянного тока. В зависимости от изучаемого свойства различают следующие видыэлектрохимических методов анализа: потенциометрию, кондуктометрию, кулонометрию, полярографию.
Потенциометрический метод анализа основан на измерении потенциалов электродов.
При погружении металла в раствор, содержащий его ионы, возможен процесс перехода ионов с поверхности металла в раствор и обратный процесс перехода ионов из раствора на поверхность металла. В результате поверхность металла приобретает заряд, знак которого зависит от того, какой из указанных процессов преобладает. На границе раздела фаз «металл – раствор» возникает разность потенциалов. Когда скорости процессов сравниваются, наступает равновесие. Электродный потенциал, соответствующий состоянию равновесия, называется равновесным потенциалом металлического электрода, обозначается буквой φ и измеряется в вольтах (В). Величина электродного потенциала зависит от природы металла, температуры и концентрации ионов металла (Mez+) в растворе (cMez+). Согласно теории Нернста значение φ при температуре 298 К рассчитывается по формуле:
φ ок/вос = φо ок/вос + 0,059/z ∙ lg cMez+ .
При погружении инертного (платинового) электрода в раствор, содержащий ионы одного химического элемента в разных степенях окисления, металлический электрод выполняет роль только передатчика электронов, на котором происходит обмен электронами окисленной и восстановленной форм системы. Указанные процессы происходят одновременно. В зависимости от природы и концентрации компонентов сначала преобладает скорость процесса перехода электронов на поверхность металла от восстановителя, либо от поверхности электрода к окислителю. Возникающий избыточный заряд за счет электростатического притяжения вызывает образование у поверхности металла двойного электрического слоя, состоящего из ионов раствора противоположного знака. Когда скорости процессов сравняются, электрод приобретает равновесный скачок потенциала, называемый окислительно–восстановительным потенциалом, значение которого рассчитывается по уравнению:
φ ок/вос = φо ок/вос + 0,059/z ∙ lg с ок / С вос.
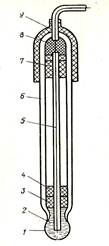
Прямая потенциометрия основана на измерении потенциала индикаторного электрода, погруженного в исследуемый раствор, и расчете концентрации определяемых ионов по уравнению Нернста. Так, для измерения рН используют стеклянный индикаторный электрод (см. рис. 51), имеющий форму пробирки с шариком на конце, сделанным из специального стекла. Электродная реакция стеклянный электрода связана с обменом ионами водорода между стеклом и раствором: Н+ (р-р) ↔ Н+(стекло).
|
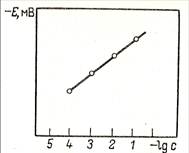
Рис. 52. Градуировочный
график.
Уравнение Нернста для стеклянного электрода при температуре 298 К имеет вид:
φ Н+/Н2 = φо Н+/Н2 - 0,059 рН,
из которого следует, что потенциал стеклянного электрода линейно зависит от рН раствора (рис. 52). Обычно стеклянный электрод функционирует в интервале рН, равном 0,5 – 12. Для измерения рН в исследуемый раствор помещают дополнительно электрод сравнения (например, хлорсеребряный), потенциал которого известен и имеет постоянное значение. Таким образом, в растворе находится система из двух электродов, ЭДС которой измеряется в милливольтах и переводится в единицы рН:
Ag, Cl│ раствор HCl │стекло│исследуемый раствор│раствор KCl │AgCl, Ag ___________________________ ______________________
стеклянный электрод электрод сравнения
Для определения концентраций других ионов применяются ионселективные электроды с относительно высокой специфичностью к отдельному иону (рNa – электрод, нитратный электрод и другие).
Потенциометрическое титрование является вариантом титриметрии, в котором конечную точку титрования устанавливают по измерению потенциала индикаторного электрода. Вблизи точки эквивалентности потенциал индикаторного электрода резко меняется. Для нахождения точки конца титрования (к.т.т.) строят кривую титрования (зависимость между потенциалом индикаторного электрода и объемом (V) титранта), как показано на рис. 53. Ординате точки перегиба кривой титрования соответствует абсцисса – объем израсходованного на титрование рабочего раствора с известной концентрацией.
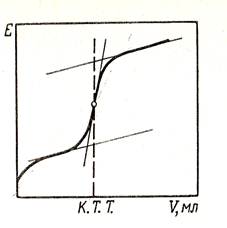
Рис. 53. Кривая потенциометрического
титрования.
По типу протекающих в растворе реакций различают кислотно-основное, окислительно-восстановительное, осадительное титрование. Индикаторный электрод выбирают в зависимости от используемой для анализа реакции.
Основными достоинствами метода являются точность, возможность проводить измерения в мутных и окрашенных растворах, где нельзя применять обычные индикаторы для титрования, а также определять несколько компонентов в растворе без предварительного их разделения и, наконец, автоматизация самого процесса титрования.
Метод полярографии предложен чешским ученым Я. Гейеровским. Метод используется для определения содержания металлов в сплавах, рудах, шлаках металлургического производства, ионы которых восстанавливаются на ртутном катоде при электролизе их растворов.
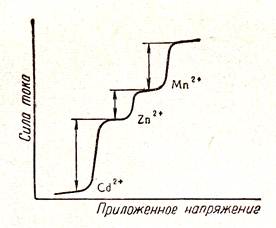
Определение основано на явлении поляризации - изменения значения потенциала электрода при пропускании через него электрического тока. При погружении в исследуемый раствор двух ртутных электродов (катод представляет собой вытекающую из капилляра по каплям ртуть, а анод – слой ртути на дне электролитической ячейки) можно наблюдать за изменением силы тока, протекающего через раствор, в зависимости от приложенного внешнего напряжения. Кривые, построенные в координатах «напряжение – ток», называются полярограммами. Они имеют неравномерный характер и напоминают волну с перегибами (рис. 54). По расположению и высоте волн можно судить о качественном составе и количественном содержании (концентрации) раствора.
Применение стандартных растворов с известной концентрацией определяемых ионов позволяет пользоваться сравнительным методом определения. По градуировочному графику зависимости силы предельного тока (высоты волны) от концентрации восстанавливающегося вещества можно легко определить неизвестную концентрацию раствора.
|
Можно использовать метод добавок, в котором сначала полярографируют исследуемый раствор, а потом приливают к нему некоторое количество стандартного раствора и снова снимают полярограмму с учетом новой концентрации раствора. По измеренной новой высоте волны из пропорции находят искомую концентрацию раствора.
Кондуктометрия – электрохимический метод анализа, основанный на измерении электропроводности растворов – способности их проводить электрический ток. Единицей измерения электропроводности является величина, обратная электрическому сопротивлению, называемая «сименсом (См)». Перенос зарядов в растворе осуществляется ионами, поэтому электропроводность зависит от их числа, т. е. концентрации раствора.
Теория метода основана на законе Кольрауша, согласно которому электропроводность раствора (χ) равна сумме электропроводностей его ионов. Так для раствора гидроксида натрия:
χ NaOH = χ Na+ + χ OH- = 217 (Cм/м).
Прямая кондуктометрия основана на измерении зависимости электропроводности растворов от их концентрации. Так как значения подвижностей различных ионов близки, селективность метода мала и при его использовании можно получить информацию только об общей концентрации ионов в растворе.
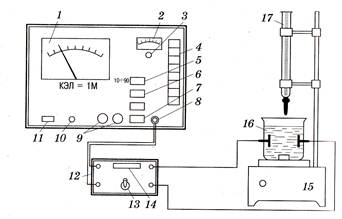
Кондуктометрическое титрование широко используется для определения точки эквивалентности в титриметрическом анализе. Оно выполняется с помощью установки, состоящей из ячейки с двумя платиновыми электродами, бюретки для раствора титранта, магнитной мешалки и прибора – кондуктометра − для измерения электропроводности (рис. 55). Измеряя электропроводность раствора после добавления небольших порций титранта, строят график зависимости в координатах «электрическая проводимость – объем титранта», по которому можно определить его эквивалентный объем. В методе используются такие химические реакции, в которых электропроводность резко меняется после достижения точки эквивалентности. К таким реакциям относятся реакции ионного обмена, в частности нейтрализация, так как подвижность ионов Н+ и ОН- выше, чем ионов металлов и анионов кислотных остатков.
|
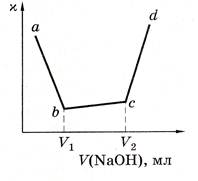
Так, при титровании сильным основанием NaOH смеси кислот (соляной и уксусной) в первую очередь со щелочью взаимодействует сильная кислота HCl, что приводит к резкому понижению электропроводности раствора (ионы Н+ связываются в молекулы воды). При титровании слабой уксусной кислоты проводимость растет, так как вместо слабой кислоты образуется соль – сильный электролит. После достижения точки эквивалентности электропроводность резко возрастает благодаря появлению в растворе избытка ионов ОН- (см. рис. 56). Объем щелочи (V1) соответствует оттитровыванию HCl, а объем (V2) – суммы кислот (HCl + СН3СООН).
|
Хроматографический анализ считается классическим методом разделения веществ. Он позволяет разделять многокомпонентные смеси газов, жидкостей и растворенных веществ методом сорбции в динамических условиях: при распределении компонентов смесей между двумя несмешивающимися фазами – подвижной (движущейся) и неподвижной. В качестве неподвижной используют твердое вещество или жидкость, нанесенную на твердую инертную подложку (носитель). Подвижной фазой служит газ или жидкость, поэтому различают газовую (ГХ) или жидкостную (ЖХ) хроматографию.
Методыхроматографического анализа классифицируют по механизму процесса и природе частиц (молекулярная, ионообменная, осадительная, распределительная хроматография) и по способам выполнения (колоночная, капиллярная, тонкослойная, бумажная хроматография).
Молекулярная хроматография основана на различной адсорбируемости молекул на адсорбентах, ионообменная – на различной способности к обмену ионов раствора. В осадительной хроматографии используется различная растворимость осадков, образуемых при взаимодействии компонентов смеси с реактивами, нанесенными на носитель. Распределительная хроматография базируется на различном распределении веществ между двумя несмешивающимися жидкостями.
Молекулярная, ионообменная или осадительная хроматография проводятся в колонках, заполненных соответственно адсорбентом, ионообменником или инертным носителем с реагентом – осадителем.
Распределительная хроматография выполняется на специальной фильтровальной бумаге (бумажная хроматография) или в тонком слое абсорбента (тонкослойная хроматография).
Хроматографическое разделение основано на том, что, перемещаясь по колонке или листу фильтровальной бумаги с различной скоростью, отдельные компоненты смеси за одинаковое время проходят различные отрезки пути.
Схема хроматографического разделения в колонке приведена на рис. 57. Исследуемую смесь компонентов А и В, растворенную в растворителе, вводят в колонку, в верхней части которой они адсорбируются. При промывании соответствующим растворителем или смесью растворителей компоненты начинают перемещаться по колонке вниз с разной скоростью, в зависимости от их адсорбционной способности. Чистые вещества собирают на выходе, где каждый компонент регистрируется с помощью различных описанных выше методов (фотоколориметрии, потенциометрии, рефрактометрии и других). Детектор с автоматической регистрацией на выходе из колонки записывает график общего вида, называемый хроматограммой (рис. 58). Каждый записанный пик соответствует отдельному компоненту смеси, а площадь пика характеризует содержание его в смеси.
Газовая хроматография используется для разделения компонентов летучих смесей. Подвижной фазой является газ (гелий, азот, водород). В зависимости от агрегатного состояния неподвижной фазы различают газотвердофазную и газожидкостную хроматографию. Разделение проводят в специальных приборах – хроматографах, куда вводят смесь с помощью микрошприца, испаряют ее при определенной температуре и вместе с газом – носителем пропускают через колонку, которая заполнена твердым адсорбентом или твердым носителем с нанесенным на нем слоем нелетучей органической жидкости. В газовых хроматографах применяют детекторы теплопроводности, измеряющие разность теплопроводности чистого газа – носителя и газа, содержащего разделяемые компоненты.
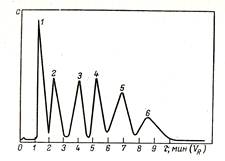
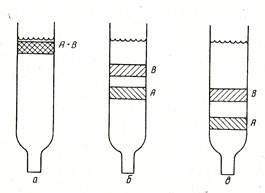
Рис. 57. Схема хроматографического Рис. 58. Хроматограмма
разделения: смеси веществ
а – ввод смеси в колонку;
б и в – элюирование
К достоинствам хроматографического метода анализа относятся быстрота и надежность, возможность одновременного определения нескольких компонентов смеси или раствора.
Выводы:
1. Использование законов электрохимии, сорбции, поглощения и отражения излучения позволило создать большое число инструментальных (физико-химических) методов анализа веществ.
2. Физико-химические методы анализа обладают следующими достоинствами: селективностью (избирательностью), которая позволяет произвести одновременно не только качественный, но и количественный анализ нескольких компонентов вещества; высокой чувствительностью; экспрессивностью (высокой скоростью проведения анализа); возможностью применения в производственных условиях для контроля технологических процессов.
Контрольные вопросы:
1. Какие методы химического анализа веществ называют инструментальными?
2. В чем состоят особенности физико-химических методов анализа?
3. Назовите основные виды взаимодействия излучения с веществами. Какие из них положены в основу оптических методов анализа?
4. В чем заключается разница между спектрофотомерией и фотоколориметрией?
5. Какие методы анализа относятся к электрохимическим?
6. Что собой представляет хроматографическая колонка? Как с ее помощью разделить смесь веществ?
Литература:
1. Коровин, Н.В. Общая химия: учеб. для технич. направл. и спец. вузов / Н.В. Коровин. – М.: Высшая школа, 2006. – §§ 16.1 – 16.3, с. 511−516.
2. Васильев, В.П. Аналитическая химия: пособие для вузов / В.П. Васильев, Р.П.Морозова, Л.А.Кочергина. – М.: Дрофа, 2004. – 416 с.
|
Дата добавления: 2014-11-16; Просмотров: 4490; Нарушение авторских прав?; Мы поможем в написании вашей работы!