КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Загальна нозологія 25 страница
|
|
|
|
Під активацією Са-каиалів розуміють їх фосфорування, у результаті чого збільшується кількість Са-каналів, здатних відкриватися при виникненні потенціалу дії. В основі фосфорування Са-каналів лежить збільшення концентрації цАМФ у саркоплазмі м'язових волокон. Усі фактори, що викликають утворення цАМФ, ведуть до активації Са-каналів. До таких, зокрема, належать:
а) катехоламіни і фармакологічні агенти, що активують fi-адренорецептори;
б) інгібітори фосфодіестерази (метилкеантини: теофілін, кофеїн та ін.). Зазначені агенти збільшують вміст цАМФ або через активацію аденілатциклази (через 0-адренорецептори), або через пригнічення руйнування цАМФ (інгібітори фосфодіестерази).
З активацією Са-каналів пов'язаний позитивний інотропний ефект катехола-мінів/Q основі його така послідовність подій: катехоламіни → активація р-адрено-рецепторів → активація аденілатциклази → утворення цАМФ → активація проте-їнкіназ → фосфорування Са-каналів сарколеми → збільшення надходження Са2+ у саркоплазму під час потенціалів дії →збільшення концентрації Са2+ у саркоплазмі → збільшення сили скорочень серця.
В умовах блокади Са-капалів зменшується надходження іонів Са в саркоплазму кардіоміоцитів і, отже, зменшується сила серцевих скорочень.
Блокаду Са-каналів викликають:
а) ендогенні фактори — дефіцит АТФ, дефіцит цАМФ, іони водню (ацидоз);
б) екзогенні фактори — двовалентні іони (Ni2+, Со2+, Мп2+), деякі тривалентні іони (La3+), органічні сполуки, що їх застосовують у практичній медицині (верапаміл, ніфедипін та ін.).
27.37. Які фактори впливають на стан систем видалення іонів кальцію з кардіоміоцитів і, як наслідок, на силу серцевих скорочень?
До таких чинників відносять:
а) серцеві глікозиди — фармакологічні препарати рослинного походження, що підвищують силу скорочень серця (препарати дигіталісу, строфантин та ін.);
б) ендогенні дигіталісоподібні і строфантиноподібні фактори. Вважають, що серцеві глікозиди, яких здавна використовують в медицині, імітують дію цих, відносно недавно відкритих, ендогенних природних факторів. Фізіологічне значення останніх сьогодні ще не встановлено.
Механізм дії серцевих глікозидів і подібних до них ендогенних факторів пов'язаний з пригніченням активності Na-K-АТФ-ази кардіоміоцитів. Наслідком цього є порушення роботи Na-K-насосів сарколеми, зменшення градієнта концентрацій іонів Na+ по обидва боки плазматичної мембрани м'язових волокон. Це призводить до порушення Na-Ca-обмінного механізму в кардіоміоцитах, у результаті чого зменшується видалення Са2+ із саркоплазми в позаклітинне середовище і збільшується внутрішньоклітинна концентрація Са2+. Останнє і обумовлює збільшення сили серцевих скорочень.
27.38. Від чого залежить сила скорочень окремих кардіоміоцитів?
На силу скорочень м'язових волокон серця впливають:
1) концентрація іонів Са2+у саркоплазмі. Залежність тут така: що вищий вміст Са2+ у саркоплазмі, то більше утворюється комплексів Са2+ з тропоніном С, то більше вивільняється центрів зв'язування (активних центрів) на актинових міофіламентах, то більше утворюється "містків" між актином і головками міозину, то більшою буде сила скорочення м'язового волокна. При зменшенні концентрації Са2+ у саркоплазмі — навпаки;
2) ступінь спорідненості тропоніну С до іонів кальцію. Іони водню й неорганічного фосфату, зв'язуючись з тропоніном С, унеможливлюють взаємодію цього білка з Са2+, у результаті чого сила скорочень кардіоміоцитів зменшується;
3) стан скорочувальних білків — актину і міозину. Велике значення має взаємне розташування актинових і міозинових міофіламентів. Воно лежить в основі залежності, що її описує закон Франка-Старлінга. При дуже сильному розтягненні м'язових волокон кількість утворюваних актоміозинових "містків" зменшується - зазначений закон не "спрацьовує", сила скорочень серця падає;
4) концентрація АТФ, енергія гідролізу якого забезпечує ковзання м'язових філамен-тів один відносно одного.
27.39. Що лежить в основі розслаблення кардіоміоцитів? Які механізми забезпечують видалення іонів кальцію із саркоплазми волокон міокарда? Що може бути причиною порушень розслаблення серцевого м'яза?
Основний процес, що визначає розслаблення кардіоміоцитів, — це видалення іонів кальцію із саркоплазми, у результаті чого концентрація Са2+ у ній зменшується і стає нижче 10"7 моль/л. При цьому комплекси Са2+ з тропоніном С розпадаються, тро-поміозин зміщується по відношенню до актинових філаментів і закриває їхні активні центри — скорочення припиняється.
Існує три механізми видалення іонів Са2 + із саркоплазми кардіоміоцитів:
1) Са-насоси плазматичної мембрани і саркоплазматичного ретикулуму. Видаляють Са2+ у позаклітинне середовище і цистерни саркоплазматичного ретикулуму. Складовою їх частиною є Са-АТФ-аза, яка для здійснення активного транспорту іонів Са2+ використовує енергію АТФ;
2) Na-Ca-обмінний механізм. Видаляє іони Са2+ у позаклітинне середовище. Є різновидом вторинного активного транспорту (антипорту). Використовує енергію градієнта концентрацій іонів натрію по обидва боки плазматичної мембрани, тому залежить від роботи Na-K-насоса, що створює цей градієнт;
3) Са-акумулятивна функція мітохондрій. Активується тільки при значному підвищенні вмісту іонів Са2+ у саркоплазмі, що найчастіше буває в умовах патології. Видалення Са2+ із саркоплазми в матрикс мітохондрій відбувається за рахунок енергії, що вивільняється в процесі транспорту електронів по дихальному ланцюгу. Використання цієї енергії на активний транспорт іонів Са2+ у мітохондрії є альтернативою окисному фосфоруванню.
Основними причинами порушень розслаблення кардіоміоцитів є:
а) дефіцит АТФ. При цьому порушується енергозабезпечення Са- і Na-K-насосів, а також не відбувається розщеплення актоміозинових "містків", що утворилися в процесі скорочення;
б) порушення роботи Са-транспортних систем. Відомі спадково обумовлені дефекти білків Са-насосів, що призводять до розвитку кардіопатії.
Порушення розслаблення кардіоміоцитів виявляють себе розвитком м'язових контрактур.
27.40. Яке значення має АТФ у забезпеченні функцій клітин міокарда? Чим можуть бути обумовлені порушення енергетичного обміну в серцевому м 'язі?
Енергія гідролізу АТФ у функціонально активних кардіоміоцитах забезпечує:
1) механічну роботу скорочень міофібрил (ковзання міофіламентів один відносно одного);
2) осмотичну роботу — активний транспорт іонів Са2+, Na +, К+ проти градієнтів концентрацій (робота іонних насосів);
3) фосфорування білків Са-каналів, фосфоламбану (білка Са-насосів саркоплазматичного ретикулуму).
Розлади енергозабезпечення кардіоміоцитів можуть бути пов'язані з порушеннями:
а) ресинтезу АТФ (гіпоксія, голодування, дефіцит вітамінів, зменшення активності ферментів енергетичного обміну);
б) транспорту АТФ із мітохондрій до місць його використання (порушення креатин-кіназної транспортної системи);
в) утилізації АТФ (зменшення АТФ-азної активності структур кардіоміоцитів).
27.41. Дайте порівняльну характеристику гіпо- і гіперкальцієвого варіантів недостатності серця.
Сучасний рівень знань про молекулярні механізми скорочувальної функції серця дозволяє виділити два принципово різних патогенетичних варіанти недостатності серця: гіпокальцієвий і гіперкальцієвий, для яких характерне відповідно зменшення і збільшення концентрації іонів Са2+ у саркоплазмі кардіоміоцитів.
Гіпокальцієвий варіант розвивається в результаті порушень збудження і електромеханічного спряження у волокнах міокарда. Це буває при аритміях (брадикардії різного походження, блокади), короткочасній ішемії міокарда (порушується фосфорування Са-каналів у результаті дефіциту АТФ), ацидозі (блокада Са-каналів іонами водню), гіпокальціємії. Виявляє себе зменшенням сили серцевих скорочень. Основний принцип лікування — підвищення вмісту іонів Са2+ у саркоплазмі кардіоміоцитів під час систоли серця. Для цього використовують: а) серцеві глікозиди; б) катехола-міни і р-адреноміметики; в) парні електричні стимули.
Гіперкальцієвий варіант розвивається в результаті посиленого надходження іонів Са2+ у саркоплазму кардіоміоцитів з позаклітинного середовища (усі види ушкодження сарколеми, при яких підвищується її проникність) або внаслідок порушень видалення Са2+ із саркоплазми (дефіцит АТФ, порушення функції Са-транспортних систем). Виявляється розвитком контрактури (перескорочення) міофібрил, у результаті чого стає неможливим розслаблення м'язових волокон, а отже, і наступне їх скорочення. Основний принцип лікування — зменшення вмісту іонів Са2+ у саркоплазмі кардіоміоцитів. Для цього використовують: а) β-адреноблокатори; б) блокатори Са-каналів.
27.42. Коли розвивається позаміокардіальна недостатність серця? Які компенсаторні механізми характерні для неї?
Позаміокардіальна недостатність розвивається в тих випадках, коли до серця притікає мало крові по венах або коли воно не в змозі прийняти всю кров, що надходить. Перше буває при гіповолемії (крововтрата) або різкому розширенні судин (колапс), друге - при накопиченні рідини в порожнині перикарда, що утруднює розширення порожнин під час діастоли.
Накопичення рідини в порожнині перикарда може відбуватися швидко і повільно. Швидке накопичення відбувається внаслідок крововиливу при пораненні або розриві серця, при перикардиті, що швидко розвивається. Через погану розтяжність перикарда в порожнині підвищується тиск, що перешкоджає діастолічному розширенню сер-
ця, виникає гостра тампонада серця. В експерименті цей процес моделюють введенням рідини в порожнину перикарда (О. Фохт). При цьому спостерігають, як зменшується кро-вонаповнення порожнин серця, знижується ударний об'єм і падає артеріальний тиск. Між внутрішньоперикардіальним і артеріальним тиском існує чітка обернена залежність: що більший внутрішньоперикардіальний тиск, то нижчий артеріальний. Венозний тиск при цьому підвищується.
Вмикання компенсаторних механізмів при перикардиті відбувається рефлекторно за участю сигналів, що надходять з трьох рецепторних полів:
1) отворів порожнистих і легеневих вен — підвищення тиску на шляхах припливу;
2) аорти і сонних синусів (синокаротидні зони) — зниження тиску на шляхах відтоку;
3) перикарда — підвищення внутрішньоперикардіального тиску. При перетинанні блукаючих і депресорних нервів, а також при вимиканні рецепторних полів за допомогою новокаїну пристосувальні реакції не здійснюються - порушення кровообігу мають набагато тяжчий перебіг. При тампонаді серця мобілізація потужних механізмів компенсації, які ведуть до посилення скорочень серця (гомео- і гетерометричні механізми, інотропний ефект катехоламінів), малоефективна або неможлива. Тому діють тільки відносно малопотужні і енергетично невигідні механізми компенсації і підтримки артеріального тиску - збільшення частоти серцевих скорочень і звуження периферичних судин. Цим і пояснюється важкий клінічний перебіг гострої тампонади серця.
При повільнішому накопиченні рідини в перикарді робота компенсаторних механізмів виявляється ефективнішою, підвищення внутрішньоперикардіального тиску протягом деякого часу може компенсуватися. Повільне накопичення рідини, що спостерігається при хронічному ексудативному перикардиті і гідроперикарді, супроводжується поступовим розтягуванням перикарда і збільшенням об'єму навколосерцевої сумки. Унаслідок цього внутрішньоперикардіальний тиск змінюється порівняно мало, а порушення кровообігу не виникають тривалий час.
27.43. Які зміни показників кардіо- і гемодинаміки характерні для недостатності серця?
При декомпенсації серця показники кардіодинаміки змінюються в такий спосіб:
1) зменшується серцевий виштовх (ударний об'єм);
2) збільшується кінцевосистолічний об 'єм;
3) збільшується кінцеводіастолічний об 'єм;
4) збільшується кінцеводіастолічний тиск, у результаті чого розвивається міогенна дилатація серця (розширення його порожнин);
5) зменшуються серцеві індекси, тобто показники, що характеризують скорочувальну активність серця. Серед них велике значення має зниження dP/dt — максимальної швидкості приросту тиску під час періоду ізоволемічного скорочення;
6) збільшується частота серцевих скорочень (тахікардія). Розвивається рефлекторно в результаті збудження рецепторів устя переповнених кров'ю порожнистих вен (рефлекс Бейнбріджа). Має також значення безпосереднє збудження клітин синусно-передсердного вузла в результаті підвищення тиску крові в порожнині правого передсердя. Розлади гемодинаміки характеризуються такими змінами:
1) зменшується хвилинний об'єм крові;
2) якщо недостатність серця розвивається за лівошлуночковим типом, то:
а) зменшується артеріальний тиску великому колі кровообігу;
б) збільшується загальний периферичний опір (реакція, спрямована на зменшення падіння артеріального тиску);
в) збільшується тиск крові в малому колі кровообігу — гіпертензія малого кола, що призводить до застою крові в легенях;
3) якщо недостатність серця розвивається за правошлуночковим типом, то:
а) зменшується артеріальний тиску малому колі кровообігу;
б) збільшується опір судин малого кола;
в) збільшується центральний венозний тиск— застій крові у великому колі кровообігу;
4) збільшується об'єм циркулюючої крові - розвивається гіперволемія. Вона є результатом затримки води в організмі і поліцитемії.
27.44. Якими клінічними синдромами й ознаками виявляє себе недостатність серця?
1. Циркуляторна гіпоксія (див. розд. 19).
2. Задишка. У її розвитку мають значення:
а) вплив надлишку іонів водню на хеморецептори судин і безпосередньо на дихальний центр;
б) набряк інтерстиціальної тканини легень і пов'язане з цим збудження J-pe-цепторів.
3. Ціаноз. Обумовлений збільшенням концентрації відновленого гемоглобіну в результаті більш повного вилучення кисню тканинами.
4. Набряки. При правошлуночковій недостатності розвиваються набряки нижньої половини тіла; при лівошлуночковій - інтерстиціальний набряк легень (синдром серцевої астми) або альвеолярний набряк (синдром набряку легень).
5. Кардіальний цироз печінки. Характерний для правошлуночкової недостатності серця. Виявляється порушеннями функції печінки й синдромом портальної гіпертензії (див. розд. 31).
6. Порушення кислотно-основного стану. Можливі такі їх варіанти:
а) негазовий ацидоз — у крові накопичуються кислі продукти обміну речовин, зокрема, молочна кислота. Є відображенням гіпоксії;
б) газовий ацидоз - може бути проявом альвеолярного набряку легень. У результаті розвитку недостатності зовнішнього дихання виникає гіперкапнія;
в) негазовий алкалоз — може бути результатом вторинного гіперальдостероніз-му і обумовленої ним гіпокаліємії;
г) газовий алкалоз — може бути наслідком рефлекторної задишки (гіпервенти-ляції), що призводить до гіпокапнії.
7. Вторинний гіперальдостеронізм (див. розд. 33). Обумовлює електролітні порушення в організмі — гіпернатріємію, гіпергідрію, гіпокаліємію.
8. Поліцитемічна гіперволемія (див. розд. 26.1).
27.45. Якими особливостями характеризується вінцевий кровообігу серці?
1. Високий рівень екстракції кисню в капілярах серця. У серці екстрагується 70—75 % 02, що надходить з артеріальною кров'ю, тимчасом як у тканинах головного мозку — 25 %, у скелетних м'язах і печінці — близько 20 %, у нирках — 10 %. Високий рівень вилучення 02 у серці пояснюється значною довжиною його капілярів і у зв'язку з цим — більшим часом контакту крові зі стінкою капілярів.
При збільшенні потреби серця в кисні вона не може бути задоволена збільшенням екстракції 02 (як у скелетних м'язах), оскільки остання і так є максимально можливою в стані спокою. Тому для забезпечення збільшених енергетичних потреб серця залишається тільки один шлях — збільшення вінцевого кровообігу.
2. Високий базальний тонус вінцевих судин. Він дає можливість у стані спокою забезпечувати вінцевий кровообіг на рівні 250-300 мл/хв, що становить близько 5 % хвилинного об'єму крові.
Високий базальний тонус вінцевих судин обумовлює високий резерв вінцевого кровообігу. Так, при зменшенні базального тонусу судин серця Інтенсивність кровообігу у них може зростати в 7—10 разів.
3. Фазний характер вінцевого кровообігу, пов'язаний з періодами серцевого циклу. Під час систоли відбувається здавлювання інтрамуральних судин - кровообіг мінімальний і становить близько 15 % загального вінцевого кровообігу. Під час діастоли здавлювання судин припиняється і кровообіг стає максимальним (близько 85 % загальної величини).
Фазність вінцевого кровообігу найбільш виражена в субендокардіальній зоні міокарда (найбільше здавлювання судин) і найменш виражена - у субепікардіальній зоні.
4. Підпорядкованість вінцевого кровообігу метаболічним потребам серця і відносна незалежність його від нервових регуляторних впливів. В умовах патології ця підпорядкованість часто порушується і збільшується чутливість вінцевих судин до нервових імпульсів.
5. Винятково висока чутливість вінцевих судин до зменшення напруги кисню в крові. Зменшення р02 артеріальної крові всього лише на 5 % істотно збільшує інтенсивність вінцевого кровообігу.
6. Недостатній розвиток колатеральних судин. При несприятливих умовах колате-ралі в серці не можуть компенсувати порушення течії крові у вінцевих судинах, тому колатеральний кровообіг тут функціонально неповноцінний.
27.46. Як здійснюється регуляція вінцевого кровообігу?
У регуляції вінцевого кровообігу розрізняють міогенну ауторегуляцію, метаболічну і нервову регуляцію.
1. Міогенна ауторегуляція. її основу становить закон Бейліса, відповідно до якого при розтягненні гладких м 'язів кровоносних судин збільшується сила їх скорочення. Міогенна ауторегуляція забезпечує сталість вінцевого кровообігу і відносну незалежність його від змін артеріального тиску. Так, при збільшенні тиску крові в аорті збільшується розтягування гладком'язових клітин вінцевих артерій, що веде до їх скорочення, підвищення тонусу артерій і збереження сталості кровообігу. При зменшенні артеріального тиску вінцевий кровообіг підтримується на постійному рівні завдяки розслабленню гладких м'язів і розширенню артерій.
Нині показано, що при розтягуванні гладком'язових клітин судин збільшується проникність їхньої плазматичної мембрани до іонів кальцію. Останні проникають у клітини і викликають їх скорочення.
2. Метаболічна регуляція. Підпорядковує вінцевий кровообіг метаболічним потребам серця. Здійснюється за допомогою цілого ряду іонів і метаболітів, серед яких іони водню, калію, молочна кислота, простагландини. Однак найбільше значення мають два фактори: зменшення напруги 02 в артеріальній крові і аденозин. Останній утворюється в результаті гідролізу аденінових нуклеотидів при гіпоксії і при посиленій роботі серця. Будучи природним блокатором Са-каналів, аденозин зменшує надходження іонів Са2+ у цитоплазму гладком'язових клітин вінцевих судин, унаслідок чого зменшується ступінь їхнього скорочення і падає базальний тонус - вінцеві судини розширюються, коронарний кровообіг зростає (рис. 129).

Рис. 129. Один з механізмів метаболічної регуляції вінцевого кровообігу
3. Нервова регуляція. Нейрогенний тонус вінцевих судин незначний, про що свідчить майже повна відсутність змін вінцевого кровообігу після повної денерва-ції судин серця. Набагато більше значення має опосередкований вплив нервової системи на коронарний кровообіг. Він здійснюється через зміни роботи серця та інтенсивності обміну речовин у ньому.
В експерименті показано можливість і безпосереднього впливу нервів на тонус вінцевих судин. Так, при подразненні парасимпатичних нервів і введенні ацетилхоліну відбувається незначне розширення вінцевих артерій. Медіатори симпатичної нервової системи (катехоламіни) при дії на а-адренорецептори викликають звуження судин, а впливаючи на р-адренорецептори — розширення. Оскільки в нормі у вінцевих судинах переважають β-адренорецептори, то загальний ефект симпатичних впливів — незначне розширення судин серця.
27.47. Що таке недостатність вінцевого кровообігу? Чим відрізняються відносна і абсолютна коронарна недостатність?
Недостатність вінцевого кровообігу - це патологічний стан, що характеризується нездатністю вінцевих судин забезпечувати кровопостачання серця відповідно до його енергетичних потреб. Інакше кажучи, виникає невідповідність між енергетичними потребами серця і доставкою кисню і поживних речовин вінцевими судинами.
Недостатність вінцевого кровообігу може бути відносною і абсолютною.
Відносна коронарна недостатність виникає у випадку первинного збільшення енергетичних потреб серця (збільшення навантаження на серце при фізичній роботі, артеріальній гіпертензії). За таких обставин інтенсивність вінцевого кровообігу може зростати, але цього буває замало, щоб задовольнити потреби серця.
Абсолютна коронарна недостатність виникає у випадку первинного порушення вінцевого кровообігу, у результаті чого зменшується доставка кисню і поживних речовин міокарду як у стані спокою, так і при збільшенні енергетичних потреб серця.
27.48. Які патогенетичні фактори можуть обумовлювати розвиток абсолютної недостатності вінцевого кровообігу?
Інтенсивність вінцевого кровообігу визначається формулою

де Q — об'ємна швидкість течії крові; Ра і Рвсн — тиск крові відповідно на початку і в кінці системи вінцевих судин; (Ра - Р) - перфузійний тиск; R - опір вінцевих судин.
Оскільки абсолютна коронарна недостатність характеризується зменшенням інтенсивності вінцевого кровообігу (зменшенням Q), можливими є два патогенетичних варіанти її розвитку.
І. Зменшення перфузійного тиску. При цьому розвивається коронарна недостатність центрального походження. її причинами можуть бути:
а) артеріальна гіпотензія (зменшення Ра п), наприклад, при всіх видах шоку, колапсі, недостатності аортальних клапанів. При зменшенні артеріального тиску спрацьовують механізми міогенної ауторегуляції вінцевого кровообігу, що підтримують його на постійному рівні. Однак, коли артеріальний тиск падає нижче 70 мм рт. ст., ці механізми виявляються неспроможними;
б) порушення венозного відтоку (збільшення Рвен). В експерименті моделюють перев'язуванням вен серця. Може мати значення при декомпенсованій недостатності правого шлуночка, коли збільшується центральний венозний і кінцеводіастолічний тиск.
II. Збільшення опору вінцевих судин. При цьому розвивається коронарна недостатність місцевого походження. Оскільки
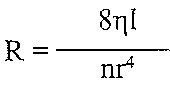
де tj — в'язкість крові; 1 — довжина судин; г — радіус судин, то збільшення опору може бути обумовлене:
а) збільшенням в 'язкості крові при порушенні її реологічних властивостей (зневоднення, поліцитемія, ДВЗ-синдром). При цьому розвиваються порушення мікроциркуляції, аж до сладж-синдрому та істинного капілярного стазу;: б) зменшенням радіуса судин. Це основний фактор розвитку абсолютної коронарної недостатності. Він викликає ішемію серця.
27.49. Які механізми можуть лежати в основі розвитку ішемії міокарда?
I. Обтураційний механізм — зменшення просвіту вінцевих артерій. Його причинами можуть бути:
а) стенозуючш атеросклероз (є причиною ішемії міокарда у 90 % випадків);
б) тромбоз вінцевих артерій (найчастіше є наслідком атеросклерозу);
в) емболія вінцевих артерій;
г) запальні процеси в стінці судин серця — коронаріїти. Бувають при ревматизмі, сифілісі.
Для розвитку клінічних ознак ішемії міокарда має значення величина "критичного стенозу ". Це мінімальне зменшення просвіту судин, при якому виникає ішемія. У людини цей показник становить 75 % (у свиней - менше, у собак - більше).
II. Ангіоспастичний механізм- спазм вінцевих судин. Його причинами можуть бути:
а) збудження а-адренорецепторів на тлі блокади р-адренорецепторів;
б) вазопресин;
в) ангіотензин II;
г) тромбоксан А^;
ґ) гіпокапнія;
д) ендотелій - біологічно активна речовина ендотеліального походження. Існує клінічна форма коронарного ангіоспазму — стенокардія Принцметала.
III. Компресійний механізм — здавлювання вінцевих судин. Може мати місце при тахікардії (збільшується загальна тривалість періоду здавлювання вінцевих судин під час систоли серця). Іноді його причиною є рубці і пухлини. В експерименті використовують для моделювання ішемії й інфаркту міокарда шляхом накладення лігатури на вінцеві артерії.
27.50. Які патогенетичні фактори впливають на міокард в умовах коронарної недостатності?
1. Гіпоксія.
2. Ацидоз. Розвивається в результаті накопичення кислих продуктів обміну речовин унаслідок порушення відтоку крові і активації гліколізу.
3. Збільшення позаклітинної концентрації іонів калію в зоні ішемії. Виявляють від самого початку порушень вінцевого кровообігу (через кілька хвилин), коли ще немає ушкодження кардіоміоцитів. У цей період з невідомих ще причин відбувається пасивний вихід іонів калію з клітин (можливо, збільшується проникність К-каналів), і його позаклітинна концентрація зростає до 8 ммоль/л і більше. Через декілька годин "витікання" іонів К+ із клітин може бути обумовлене порушенням роботи Na-K-насосів (дефіцит АТФ) та збільшенням проникності ушкоджених мембран.
27.51. Які наслідки для міокарда може мати недостатність вінцевого кровообігу?
1. Порушення скорочувальної здатності міокарда з розвитком недостатності серця.
2. Поява аномальної електричної активності — електрична нестабільність серця, розвиток аритмій.
3. Ушкодження кардіоміоцитів, обумовлене ішемією (див. розд. 11).
4. Реперфузійний синдром.
27.52. Які порушення скорочувальної функції міокарда можуть виникати при його ішемії?
При ішемії міокарда розрізняють ранні і пізні порушення його скорочувальної функції.
Ранні порушення виникають дуже рано, вже при незначному дефіциті АТФ. Вони є відображенням гіпокальцієвого варіанта порушень скорочувальної функції серцевого м'яза і розвиваються в результаті блокади Са-каналів сарколеми кардіоміоцитів. Порушення провідності Са-каналів в умовах ішемії має щонайменше два механізми:
а) дефіцит АТФ → порушення фосфорування білків Са-каналів;
б) активація гліколізу →накопичення в клітинах іонів Н+ → безпосередня блокада Са-каналів.
Наслідком зазначених порушень є зменшення надходження іонів Са2+ у саркоплазму м'язових волокон, розлади електромеханічного спряження, зменшення сили скорочень кардіоміоцитів.
Пізні порушення скорочувальної функції виникають при тривалих розладах вінцевого кровообігу - понад ЗО хв. їх відносять до гіперкальцієвого (контрактурного) типу порушень. Концентрація іонів кальцію в саркоплазмі кардіоміоцитів збільшується через дві основні причини:
а) порушення видалення іонів Са2+ із саркоплазми внаслідок дефіциту АТФ;
б) збільшення надходження іонів Са2+ у м'язові волокна через ушкоджену плазматичну мембрану клітин.
У результаті збільшення вмісту Са2+ порушуються процеси розслаблення кардіоміоцитів, настає контрактура міофібрил, порушується скорочувальна здатність серця.
27.53. Які механізми можуть обумовлювати розвиток аритмій при ішемії міокарда?
Розвиток аритмії пов'язаний з електричною нестабільністю серця, що виникає вже через кілька хвилин від початку розвитку ішемії міокарда. Основна її причина — вихід іонів калію з кардіоміоцитів, у результаті чого збільшується їх позаклітинна концентрація. Це викликає часткову деполяризацію сарколеми клітин, що перебувають поруч. Залежно від рівня підвищення вмісту позаклітинного калію можливі такі наслідки:
|
|
|
|
|
Дата добавления: 2014-12-07; Просмотров: 399; Нарушение авторских прав?; Мы поможем в написании вашей работы!