КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Образование мевалоната
|
|
|
|
Сложный путь синтеза холестерола можно разделить на 3 этапа
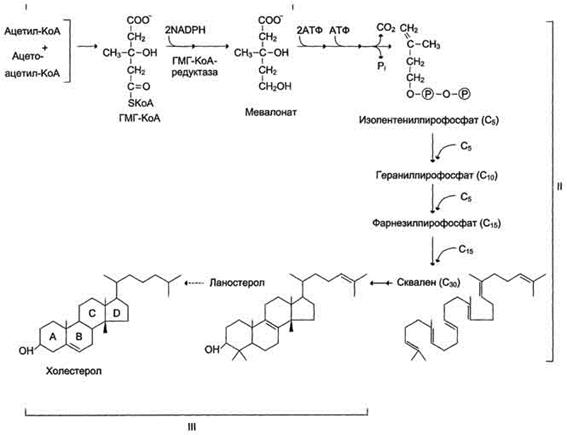
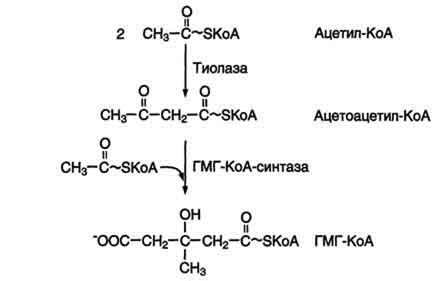
Первый этап заканчивается образованием мевалоната (мевалоновой кислоты). Две молекулы ацетил-КоА конденсируются ферментом тиолазой с образованием ацетоацетил-КоА. Фермент щдроксиметилглутарил-КоА-синтаза присоединяет третий ацетильный остаток с образованием ГМГ-КоА (3-гидрокси-3-метилглутарил-КоА). Эта последовательность реакций сходна с начальными стадиями синтеза кетоновых тел:
Следующая реакция, катализируемая ГМГ-КоА-редуктазой, является регуляторной в метаболическом пути синтеза холестерола. В этой реакции происходит восстановление ГМГ-КоА до мевалоната с использованием 2 молекул NADPH. Фермент ГМГ-КоА-редуктаза - гликопротеин, пронизывающий мембрану ЭР, активный центр которого выступает в цитозоль.
Образование сквалена На втором этапе синтеза мевалонат превращается в пятиуглеродную изопреноидную структуру, содержащую пирофосфат - изопентенилпирофосфат. Продукт конденсации 2 изопреновых единиц - геранилпирофосфат. Присоединение ещё 1 изопреновой единицы приводит к образованию фарнезилпирофосфата - соединения, состоящего из 15 углеродных атомов. Две молекулы фарнезилпирофосфата конденсируются с образованием сквалена - углеводорода линейной структуры, состоящего из 30 углеродных атомов.
Образование холестерола. На третьем этапе синтеза холестерола сквален через стадию образования эпоксида ферментом циклазой превращается в молекулу ланостерола, содержащую 4 конденсированных цикла и 30 атомов углерода. Далее происходит 20 последовательных реакций, превращающих ланостерол в холестерол. На последних этапах синтеза от ланостерола отделяется 3 атома углерода, поэтому холестерол содержит 27 углеродных атомов.У холестерола имеется насыщенная разветвлённая боковая цепь из 8 углеродных атомов в положении 17, двойная связь в кольце В между атомами углерода в положениях 5 и 6, а также гидроксильная группа в положении 3.В организме человека изопентенилпирофосфат также служит предшественником убихинона (KoQ) и долихола, участвующего в синтезе гликопротеинов.
Этерификация холестерола. В некоторых тканях гидроксильная группа холестерола этерифицируется с образованием более гидрофобных молекул - эфиров холестерола. Реакция катализируется внутриклеточным ферментом АХАТ (ацилКоА:холестеролаиилтрансферазой). Реакция этерификации происходит также в крови в ЛПВП, где находится фермент ЛХАТ (лецитин:холестеролацилтрансфераза). Эфиры холестерола - форма, в которой они депонируются в клетках или транспортируются кровью. В крови около 75% холестерола находится в виде эфиров.
71.Синтез желчных кислот из холестерина. Конъюгация желчных кислот, первичные и вторичные желчные кислоты. Выведение желчных кислот и холестерина из организма.
Жёлчные кислоты синтезируются в печени из холестерола. Часть жёлчных кислот в печени подвергается реакции конъюгации - соединения с гидрофильными молекулами (глицином и таурином). Жёлчные кислоты обеспечивают эмульгирование жиров, всасывание продуктов их переваривания и некоторых гидрофобных веществ, поступающих с пищей, например жирорастворимых витаминов и холестерола. Жёлчные кислоты также всасываются, через юротную вену попадают опять в печень и многократно используются для эмульгирования жиров. Этот путь называют энтерогепатической циркуляцией жёлчных кислот.
Синтез жёлчных кислот. В организме за сутки синтезируется 200- 600 мг жёлчных кислот. Первая реакция синтеза – образование 7-α-гидроксихолестерола - является регуляторной. Фермент 7-α-гидроксилаза, катализирующий эту реакцию, ингибируется конечным продуктом - жёлчными кислотами. 7-α-Гидроксилаза представляет собой одну из форм цитохрома Р450 и использует кислород как один из субстратов. Один атом кислорода из О2 включается в гидроксильную группу в положении 7, а другой восстанавливается до воды. Последующие реакции синтеза приводят к формированию 2 видов жёлчных кислот: холевой и хенодезоксихолевой, которые называют "первичными жёлчными кислотами".
Конъюгирование жёлчных кислот. Конъюгирование - присоединение ионизированных молекул глицина или таурина к карбоксильной группе жёлчных кислот; усиливает их детергентные свойства, так как увеличивает амфифильность молекул. Конъюгация происходит в клетках печени и начинается с образования активной формы жёлчных кислот - производных КоА. Затем присоединяется таурин или глицин, и в результате образуется 4 варианта конъюгатов: таурохолевая и таурохенодезоксихолевая, гликохолевая или гликохенодезоксихолевая кислоты (они значительно более сильные эмульгаторы, чем исходные жёлчные кислоты). Конъюгатов с глицином образуется в 3 раза больше, чем с таурином, так как количество таурина ограничено.
Энтерогепатическая циркуляция жёлчных кислот. Превращения жёлчных кислот в кишечнике. Продукты гидролиза жиров всасываются в основном в верхнем отделе тонкого кишечника, а соли жёлчных кислот - в подвздошной кишке. Около 95% жёлчных кислот, попавших в кишечник, возвращается в печень через воротную вену, затем опять секретируются в жёлчь и повторно используются в эмульгировании жиров. Этот путь жёлчных кислот называют энтерогепатической циркуляцией. В сутки всего реабсорбируется 12-32 г солей жёлчных кислот, так как в организме имеется 2-4 г жёлчных кислот, и каждая молекула жёлчной кислоты проходит этот крут 6-8 раз. Часть жёлчных кислот в кишечнике подвергается действию ферментов бактерий, которые отщепляют глицин и таурин, а также гидроксильную группу в положении 7 жёлчных кислот. Жёлчные кислоты, лишённые этой гидроксильной группы, называют вторичными. Вторичные жёлчные кислоты: дезоксихолевая, образующаяся из холевой, и литохолевая, образующаяся из дезоксихолевой, хуже растворимы, медленнее всасываются в кишечнике, чем первичные жёлчные кислоты. Поэтому с фекалиями в основном удаляются вторичные жёлчные кислоты. Однако реабсорбированные вторичные жёлчные кислоты в печени опять превращаются в первичные и участвуют в эмульгировании жиров. За сутки из организма выводится 500-600 мг жёлчных кислот. Путь выведения жёлчных кислот одновременно служит и основным путём выведения холестерола из организма. Для восполнения потери жёлчных кислот с фекалиями в печени постоянно происходит синтез жёлчных кислот из холестерола в количестве, эквивалентном выведенным жёлчным кислотам. В результате пул жёлчных кислот (2-4 г) остаётся постоянным.
Выведение холестерола из организма. Структурная основа холестерола - кольца циклопентанпергидрофенантрена - не может быть расщеплена до СО2 и воды, как другие органические компоненты, поступающие с пищей или синтезированные в организме. Поэтому основное количество холестерола выводится в виде жёлчных кислот.
Некоторое количество жёлчных кислот выделяется в неизменённом виде, а часть подвергается действию ферментов бактерий в кишечнике. Продукты их разрушения (в основном, вторичные жёлчные кислоты) выводятся из организма.
Часть молекул холестерола в кишечнике под действием ферментов бактерий восстанавливается по двойной связи в кольце В, в результате чего образуютря 2 типа молекул - холестанол и копростанол, выводимые с фекалиями. В сутки из организма выводится от 1,0 г до 1,3 г холестерола, основная часть удаляется с фекалиями,
72.ЛПНП и ЛПВП - транспортные, формы холестерина в крови, роль в обмене холестерина. Гиперхолестеринемия. Биохимические основы развития атеросклероза.
Холестерол транспортируется кровью только в составе ЛП. ЛП обеспечивают поступление в ткани экзогенного холестерола, определяют потоки холестерола между органами и выведение избытка холестерола из организма.
Транспорт экзогенного холестерола. Холестерол поступает с пищей в количестве 300-500 мг/сут, в основном в виде эфиров. После гидролиза, всасывания в составе мицелл, этерификации в клетках слизистой оболочки кишечника эфиры холестерола и небольшое количество свободного холестерола включаются в состав ХМ и поступают в кровь. После удаления жиров из ХМ под действием ЛП-липазы холестерол в составе остаточных ХМ доставляется в печень. Остаточные ХМ взаимодействуют с рецепторами клеток печени и захватываются по механизму эндоцитоза. Затем ферменты лизосом гидролизуют компоненты остаточных ХМ, и в результате образуется свободный холестерол. Экзогенный холестерол, поступающий таким образом в клетки печени, может ингибировать синтез эндогенного холестерола, замедляя скорость синтеза ГМГ-КоА-редуктазы.
Транспорт эндогенного холестерола в составе ЛПОНП (пре-β-липопротеинов). Печень - основное место синтеза холестерола. Эндогенный холестерол, синтезированный из исходного субстрата ацетил-КоА, и экзогенный, поступивший в составе остаточных ХМ, образуют в печени общий фонд холестерола. В гепатоцитах триацилглицеролы и холестерол упаковываются в ЛПОНП. В их состав входят, кроме того, апопротеин В-100 и фоефолипиды. ЛПОНП сек-ретируются в кровь, где получают от ЛПВП апопротеины Е и С-IIВ крови на ЛПОНП действует ЛП-липаза, которая, как и в ХМ, активируется апоС-II гидролизует жиры до глицерола и жирных кислот. По мере уменьшения количества ТАГ в составе ЛПОНП они превращаются в ЛППП. Когда количество жиров в ЛППП уменьшается, апопротеины С-II реносятся обратно на ЛПВП. Содержание холестерола и его эфиров в ЛППП достигает 45%; часть этих липопротеинов захватывается клетками печени через рецепторы ЛПНП, которые взаимодействуют и с апоЕ и с апоВ-100.
Транспорт холестерола в составе ЛПНП. Рецепторы ЛПНП. На ЛППП, оставшиеся в крови, продолжает действовать ЛП-липаза, и они превращаются в ЛПНП, содержащие до 55% холестерола и его эфиров. Апопротеины Е и С-II реносятся обратно в ЛПВП. Поэтому основным апопротеином в ЛПНП служит апоВ-100. Апопротеин В-100 взаимодействует с рецепторами ЛПНП и таким образом определяет дальнейший путь холестерола. ЛПНП - основная транспортная форма холестерола, в которой он доставляется в ткани. Около 70% холестерола и его эфиров в крови находится в составе ЛПНП. Из крови ЛПНП поступают в печень (до 75%) и другие ткани, которые имеют на своей поверхности рецепторы ЛПНП. Рецептор ЛПНП - сложный белок, состоящий из 5 доменов и содержащий углеводную часть. Рецепторы ЛПНП синтезируются в ЭР и аппарате Гольджи, а затем экспонируются на поверхности клетки, в специальных углублениях, выстланных белком клатрином. Эти углубления называют окаймлёнными ямками. Выступающий на поверхность N-концевой домен рецептора взаимодействует с белками апоВ-100 и апоЕ; поэтому он может связывать не только ЛПНП, но и ЛППП, ЛПОНП, остаточные ХМ, содержащие эти апопротеины. Клетки тканей содержат большое количество рецепторов ЛПНП на своей поверхности: например, на одной клетке фибробласта имеется от 20 000 до 50 000 рецепторов. Из этого следует, что холестерол поступает в клетки из крови в основном в составе ЛПНП. Если количество холестерола, поступающего в клетку, превышает её потребность, то синтез рецепторов ЛПНП подавляется, что уменьшает поток холестерола из крови в клетки. При снижении концентрации свободного холестерола в клетке, наоборот, активируется синтез ГМГ-КоА-редуктазы и рецепторов ЛПНП. В регуляции синтеза рецепторов ЛПНП участвуют гормоны: инсулин и трийодтиронин (Т3), полрвые гормоны. Они увеличивают образование рецепторов ЛПНП, а глюкокортикоиды (в основном кортизол) уменьшают. Эффекты инсулина и Т3, вероятно, могут объяснить механизм гиперхолестеролемии и увеличение риска атеросклероза при сахарном диабете или гипотиреозе.
Роль ЛПВП в обмене холестерола. ЛПВП выполняют 2 основные функции: они поставляют апопротеины другим ЛП в крови и участвуют в так называемом "обратном транспорте холестерола". ЛПВП синтезируются в печени и в небольшом количестве в тонком кишечнике в виде "незрелых липопротеинов" - предшественников ЛПВП. Они имеют дисковидную форму, небольшой размер и содержат высокий процент белков и фосфолипидов. В печени в ЛПВП включаются апопротеины А, Е, С-II, фермент ЛХАТ. В крови апоС-II и апоЕ переносятся с ЛПВП на ХМ и ЛПОНП. Предшественники ЛПВП пракгически не содержат холестерола и ТАГ и в крови обогащаются холестеролом, получая его из других ЛП и мембран клеток. Для переноса холестерола в ЛПВП существует сложный механизм. На поверхности ЛПВП находится фермент ЛХАТ - лецитишхолестерол-ацилтрансфераза. Этот фермент превращает холестерол, имеющий гидроксильную группу, выступающую на поверхность липопротеинов или мембран клеток, в эфиры холестерола. Радикал жирной кислоты переносится от фосфатидилхолита (лецитина) на гидроксильную группу холестерола. Реакция активируется апопротеином A-I, входящим в состав ЛПВП. Гидрофобная молекула, эфира холестерола перемещается внутрь ЛПВП. Таким образом, частицы ЛПВП обогащаются эфирами холестерола. ЛПВП увеличиваются в размерах, из дисковидных небольших частиц превращаются в частицы сферической формы, которые называют ЛПВП3, или "зрелые ЛПВП". ЛПВП3 частично обменивают эфиры холестерола на триацилглицеролы, содержащиеся в ЛПОНП, ЛППП и ХМ. В этом переносе участвует " белок, переносящий эфиры холестерина" (он также называется aпoD). Таким образом, часть эфиров холестерола переносится на ЛПОНП, ЛППП, а ЛПВП3 за счёт накопления триацилглицеролов увеличиваются в размерах и превращаются в ЛПВП2. ЛПОНП под действием ЛП-липазы превращаются сначала в ЛППП, а затем в ЛПНП. ЛПНП и ЛППП захватываются клетками через рецепторы ЛПНП. Таким образом, холестерол из всех тканей возвращается в печень в основном в составе ЛПНП, но в этом участвуют также ЛППП и ЛПВП2. Практически весь холестерол, который должен быть выведен из организма, поступает в печень и уже из этого органа выделяется в виде производных с фекалиями. Путь возвращения холестерола в печень называют "обратным транспортом" холестерола.
Гиперхолестеролемия. Роль алиментарных факторов в развитии гиперхолестеролемии. Концентрация холестерола в крови взрослых людей составляет 200±50 мг/дл (5,2±1,2 ммоль/л) и, как правило, увеличивается с возрастом. Превышение нормальной концентрации холестерола в крови называют гиперхолестеролемией. Гиперхолестеролемия часто развивается вследствие избыточного поступления холестерола с пищей, а также углеводов и жиров. Гиперкалорийное питание - один из распространённых факторов развития гиперхолестеролемии, так как для синтеза холестерола необходимы только ацетил-КоА, АТФ и NADPH. Все эти субстраты образуются при окислении глюкозы и жирных кислот, поэтому избыточное поступление этих компонентов пищи способствует развитию гиперхолестеролемии. В норме поступление холестерола с пищей снижает синтез собственного холестерола в печени, однако с возрастом эффективность регуляции у многих людей снижается. Правильное питание в течение всей жизни - важнейший фактор профилактики гаперхолестеролемии. Доказана корреляция между увеличением концентрации холестерола в плазме крови и смертностью от заболеваний ССС - инфаркта миокарда и инсульта, развивающихся в результате атеросклероза.
Ген рецептора ЛПНП: структура и типы мутаций. Наследственные факторы играют важную роль в предрасположенности к развитию атеросклероза. Наиболее часто встречаются мутации в структуре гена рецептора ЛПНП. Ген рецептора ЛПНП находится в хромосоме 19 и состоит из 18 экзонов. Различные группы экзонов кодируют различные домены в составе этого белка. Мутации в этом гене подробно изучены и разделены на 4 класса. Первый класс мутаций, наиболее распространённый, приводит к полному отсутствию рецептора; второй класс мутаций характеризуется тем, что рецептор синтезируется, но не может транспортироваться на поверхность клетки; третий класс мутаций соответствует ситуации, когда рецептор транспортируется на поверхность клеток, но не связывает ЛПНП; четвёртый класс мутаций - рецептор связывает ЛПНП, но не происходит эндоцитоз. Изменения структуры рецепторов ЛПНП в результате всех типов мутаций приводит к гиперхолестеролемии; так как ЛПНП не захватываются клетками, и холестерол в составе ЛПНП накапливается в крови.
Семейная гиперхолестеролемия. Любой дефект рецептора ЛПНП или белка апоВ-100, взаимодействующего с ним, приводит к развитию наиболее распространённого наследственного заболевания - семейной гиперхолестеролемии. Причиной этого аутосомно-доминантного заболевания выступают указанные выше мутации в гене рецептора ЛПНП. Гетерозиготы, имеющие один нормальный ген, а другой дефектный, встречаются с частотой 1:500 человек, у некоторых народностей Африки - даже 1:100 человек. Количество рецепторов ЛПНП на поверхности клеток у гетерози-гот снижено вдвое, а концентрация холестерола в плазме, соответственно, вдвое повышается. У гетерозигот концентрация холестерола в крови в 35-40 лет достигает 400-500 мг/дл, что приводит к выраженному атеросклерозу и ранней смерти в результате инфаркта миокарда или инсульта. Гомозиготы встречаются редко - 1:1 000 000 человек. Концентрации холестерола и ЛПНП в крови таких больных уже в раннем детском возрасте увеличены в 5-6 раз. ЛПНП захватываются макрофагами путём фагоцитоза. Макрофаги, нагруженные избытком холестерола и других лигшдов, содержащихся в ЛПНП, откладываются в коже и даже сухожилиях, образуя так называемые ксантомы. Холестерол откладывается также и в стенках артерий, образуя атеросклеротические бляшки. Такие дети без экстренных мер лечения погибают в возрасте 5-6 лет. Лечение данной формы заболевания проводят путём удаления ЛПНП из крови с помощью плазмафереза, но наиболее радикальный метод лечения - трансплантация печени. Печень донора с нормальным количеством рецепторов ЛПНП существенно понижает концентрацию холестерола в крови и предотвращает раннюю смерть от атеросклероза. Кроме генетических дефектов рецептора ЛПНП, причинами гиперхолестеролемии и, следовательно, атеросклероза являются наследственные дефекты в структуре апоВ-100, а также повышенные синтез или секреция апоВ-100 в случае семейной комбинированной гиперли-пидемии, при которой в крови повышены концентрации и холестерола и триацилглицеролов.
Молекулярные механизмы патогенеза атеросклероза
Развитие атеросклероза проходит несколько стадий
Процесс начинается с повреждения эндотелия сосудов, причём повреждение может иметь различные механизмы. Важнейший механизм - повреждение эндотелия за счёт изменённой структуры ЛПНП, например в результате активации свободнорадикального ПОЛ в составе ЛПНП; повреждение провоцируется свободными радикалами, образующимися в процессе метаболизма или поступающими извне. В ходе ПОЛ в ЛПНП изменяется не только структура самих липидов, но и нарушается структура апопротеинов. Окисленные ЛПНП захватываются макрофагами через скевенджер-рецепторы. Этот процесс не регулируется количеством поглощённого холестерола, как в случае его поступления в клетки через специфические рецепторы, поэтому макрофаги перегружаются холестеролом и превращаются в "пенистые клетки", которые проникают в субэндотелиальное пространство. Это приводит к образованию жировых полосок в стенке кровеносных сосудов. На этой стадии эндотелий сосудов может сохранять свою структуру. При увеличении количества "пенистых клеток" происходит повреждение эндотелия сосудов. В норме клетки эндотелия секретируют простагландин I2 (простациклин I2), который ингибирует агрегацию тромбоцитов. При повреждении клеток эндотелия тромбоциты активируются. Во-первых, они секретируют тромбоксан А2 (ТХ А2, который стимулирует агрегацию тромбоцитов, что может привести к образованию тромба в области атеросклеротической бляшки; во-вторых, тромбоциты начинают продуцировать пептид - тромбоцитарный фактор роста, стимулирующий пролиферацию ГМК. ГМК мигрируют из медиального слоя во внутренний слой артериальной стенки и способствуют таким образом росту бляшки. Далее происходит прорастание бляшки фиброзной тканью (коллагеном ластином); клетки под фиброзной оболочкой некротизируются, а холестерол откладывается в межклеточном пространстве. На этой стадии в центре бляшки образуются даже холестериновые кристаллы. На последних стадиях развития бляшка пропитывается солями кальция и становится очень плотной. В области бляшки часто образуются тромбы, перекрывающие просвет сосуда, что приводит к острому нарушению кровообращения в соответствующем участке ткани и развитию инфаркта. Чаще всего атеросклеротические бляшки развиваются в артериях миокарда, поэтому наиболее распространённое заболевание, развивающееся в результате атеросклероза, - инфаркт миокарда.
73. Механизм возникновения желчнокаменной болезни (холестериновые камни). Применение хенодезокеихолевой кислоты для лечения желчнокаменной болезни.
Желчнокаменная болезнь - патологический процесс, при котором в жёлчном пузыре образуются камни, основу которых составляет холестерол.
Выделение холестерола в жёлчь должно сопровождаться пропорциональным выделением жёлчных кислот и фосфолипидов, удерживающих гидрофобные молекулы холестерола в жёлчи в мицеллярном состоянии У большинства больных желчнокаменной болезнью активность ГМГ-КоА-редуктазы повышена, следовательно увеличен синтез холестерола, а активность 7-α-гидроксилазы, участвующей в синтезе жёлчных кислот, снижена. В результате синтез холестерола увеличен, а синтез жёлчных кислот из него замедлен, что приводит к диспропорции количества холестерола и жёлчных кислот," секретируемых в жёлчь.
Если эти пропорции нарушены, то холестерол начинает осаждаться в жёлчном пузыре, образуя вначале вязкий осадок, который постепенно становится более твёрдым. Иногда он пропитывается билирубином - продуктом распада тема, белками и солями кальция. Камни, образующиеся в жёлчном пузыре, могут состоять только из холестерола (холестериновые камни) или из смеси холестерола, билирубина, белков и кальция. Холестериновые камни обычно белого цвета, а смешанные камни - коричневого цвета разных оттенков. Причин, приводящих к изменению соотношения жёлчных кислот и холестерола, в жёлчи много: пища, богатая холестеролом, гиперкалорийное питание, застой жёлчи в жёлчном пузыре, нарушение энтерогепатической циркуляции, нарушения синтеза жёлчных кислот, инфекции жёлчного пузыря. Если камни начинают перемещаться из жёлчного пузыря в жёлчные протоки, то они вызывают спазм жёлчного пузыря и протоков, что больной ощущает как приступ сильной боли. Если камень перекрывает проток некоторое время, то нарушается поступление жёлчи в кишечник, жёлчные пигменты проходят через мембраны гепатоцитов в сторону синусоидов и попадают в кровь, что приводит к развитию об-турационной (подпечёночной желтухи).
Лечение желчнокаменной болезни. В начальной стадии образования камней можно применять в качестве лекарства хенодезоксихолевую кислоту. Попадая в жёлчный пузырь, эта жёлчная кислота постепенно растворяет осадок холестерола (холестериновые камни), однако это медленный процесс, требующий нескольких месяцев.
74. Общая схема источников и путей расходования аминокислот в тканях. Динамическое состояние белков в организме.
Значение аминокислот для организма в первую очередь определяется тем, что они используются для синтеза белков, метаболизм которых занимает особое место в процессах обмена веществ между организмом и внешней средой. Объясняется это тем, что белки входят во все основные структурные компоненты клеток, тканей и органов тела человека и животных, выполняют ферментативные функции, участвуют в переносе веществ через мембраны и т.д. Важную роль в координации работы всех систем клеток играют белковые гормоны. Аминокислоты непосредственно участвуют в биосинтезе не только белков, но и большого количества других биологически активных соединений, регулирующих процессы обмена веществ в организме, таких как нейромедиаторы и гормоны - производные аминокислот. Аминокислоты служат донорами азота при синтезе всех азотсодержащих небелковых соединений, в том числе нуклео-тидов, тема, креатина, холина и других веществ. Катаболизм аминокислот может служить источником энергии для синтеза АТФ. Энергетическая функция аминокислот становится значимой при голодании, некоторых патологических состояниях (сахарный диабет и др.) и преимущественно белковом питании. Именно обмен аминокислот осуществляет взаимосвязь многообразных химических превращений в живом организме. Фонд свободных аминокислот организма составляет примерно 35 г. Содержание свободных аминокислот в крови в среднем равно 35-65 мг/дл. Большая часть аминокислот входит в состав белков, количество которых в организме взрослого человека нормального телосложения составляет примерно 15 кг. Источники свободных аминокислот в клетках - белки пищи, собственные белки тканей и синтез аминокислот из углеводов. Многие клетки, за исключением высокоспециализированных (например, эритроцитов), используют аминокислоты для синтеза белков, а также большого количества других веществ: фосфолипидов мембран, гема, пуриновых и пиримидиновых нуклеотидов, биогенных аминов (катехоламинов, гистамина) и других соединений. Какой-либо специальной формы депонирования аминокислот, подобно глюкозе (в виде гликогена) или жирных кислот (в виде триацилглицеролов), не существует. Поэтому резервом аминокислот могут служить все функциональные и структурные белки тканей, но преимущественно белки мышц, поскольку их больше, чем всех остальных. В организме человека в сутки распадается на аминокислоты около 400 г белков, примерно такое же количество синтезируется. Поэтому тканевые белки не могут восполнять затраты аминокислот при их катаболизме и использовании на синтез других веществ. Первичными источниками аминокислот не могут служить и углеводы, так как из них синтезируются только углеродная часть молекулы большинства аминокислот, а аминогруппа поступает от других аминокислот. Следовательно, основным источником аминокислот организма служат белки пищи.
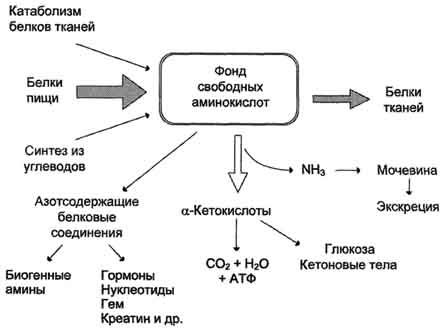
Аминокислоты (свободные и в составе белков) содержат почти 95% всего азота, поэтому именно они поддерживают азотистый баланс организма. Азотистый баланс - разница между количеством азота, поступающего с пищей, и количеством выделяемого азота (преимущественно в виде мочевины и аммонийных солей). Если количество поступающего азота равно количеству выделяемого, то наступает азотистое равновесие. Такое состояние бывает у здорового человека при нормальном питании. Азотистый баланс может быть положительным (азота поступает больше, чем выводится) у детей, а также у пациентов, выздоравливающих после тяжёлых болезней. Отрицательный азотистый баланс (выделение азота преобладает над его поступлением) наблюдают при старении, голодании и во время тяжёлых заболеваний. При безбелковой диете азотистый баланс становится отрицательным. Соблюдение подобной диеты в течение недели приводит к тому, что количество выделяемого азота перестаёт увеличиваться и стабилизируется примерно на величине 4 г/сут. Такое количество азота содержится в 25 г белка. Значит, при белковом голодании в сутки в организме расходуется около 25 г собственных белков тканей. Минимальное количество белков в пище, необходимое для поддержания азотистого равновесия, соответствует 30-50 г/cyт, оптимальное же количество при средней физической нагрузке составляет ∼100-120 г/сут.
75. Переваривание белков. Протеиназы - пепсин, трипсин, химотрипсин; проферменты протеиназ и механизмы их превращения в ферменты. Субстратная специфичность протеиназ. Экзопептидазы и эндопептидазы.
В пищевых продуктах содержание свободных аминокислот очень мало. Подавляющее их количество входит в состав белков, которые гидролизуются в ЖКТ под действием ферментов протеаз (пептидщцролаз). Субстратная специфичность этих ферментов заключается в том, что каждый из них с наибольшей скоростью расщепляет пептидные связи, образованные определёнными аминокислотами. Протеазы, гидролизующие пептидные связи внутри белковой молекулы, относят к группе эндопептидаз. Ферменты, относящиеся к группе экзопептидаз, гидролизуют пептидную связь, образованную концевыми аминокислотами. Под действием всех протеаз ЖКТ белки пищи распадаются на отдельные аминокислоты, которые затем поступают в клетки тканей.
Переваривание белков в желудке
Желудочный сок - продукт нескольких типов клеток. Обкладочные (париетальные) клетки стенок желудка образуют соляную кислоту, главные клетки секретируют пепсиноген. Добавочные и другие клетки эпителия желудка выделяют муцинсодержащую слизь. Париетальные клетки секретируют в полость желудка также гликопротеин, который называют "внутренним фактором" (фактором Касла). Этот белок связывает "внешний фактор" - витамин В12, предотвращает его разрушение и способствует всасыванию.
Образование и роль соляной кислоты. Основная пищеварительная функция желудка заключается в том, что в нём начинается переваривание белка. Существенную роль в этом процессе играет соляная кислота. Белки, поступающие в желудок, стимулируют выделение гистамина и группы белковых гормонов - гастринов которые, в свою очередь, вызывают секрецию НСI и профермента - пепсиногена. Источником Н+ является Н2СО3, которая образуется в обкладочных клетках желудка из СО2, диффундирующего из крови, и Н2О под действием фермента карбоангидразы (карбонатдегидра-тазы):
Н2О + СО2 → Н2СО3 → НСО3- + H+
Диссоциация Н2СО3 приводит к образованию бикарбоната, который с участием специальных белков выделяется в плазму в обмен на С1-, и ионов Н+, которые поступают в просвет желудка путём активного транспорта, катализируемого мембранной Н+/К+-АТФ-азой. При этом концентрация протонов в просвете желудка увеличивается в 106 раз. Ионы Сl- поступают в просвет желудка через хлоридный канал. Концентрация НСl в желудочном соке может достигать 0,16 М, за счёт чего значение рН снижается до 1,0-2,0. Приём белковой пищи часто сопровождается выделением щелочной мочи за счёт секреции большого количества бикарбоната в процессе образования НСl. Под действием НСl происходит денатурация белков пищи, не подвергшихся термической обработке, что увеличивает доступность пептидных связей для протеаз. НСl обладает бактерицидным действием и препятствует попаданию патогенных бактерий в кишечник. Кроме того, соляная кислота активирует пепсиноген и создаёт оптимум рН для действия пепсина.
Механизм активации пепсина. Под действием гастринов в главных клетках желудочных желёз стимулируются синтез и секреция пепсиногена - неактивной формы пепсина. Пепсиноген - белок, состоящий из одной полипептидной цепи с молекулярной массой 40 кД. Под действием НСl он превращается в активный пепсин (молекулярная масса 32,7 кД) с оптимумом рН 1,0-2,5. В процессе активации в результате частичного протеолиза от N-конца молекулы пепсиногена отщепляются 42 аминокислотных остатка, которые содержат почти все положительно заряженные аминокислоты, имеющиеся в пепсиногене. Таким образом, в активном пепсине преобладающими оказываются отрицательно заряженные аминокислоты, которые участвуют в конформационных перестройках молекулы и формировании активного центра. Образовавшиеся под действием НСl активные молекулы пепсина быстро активируют остальные молекулы пепсиногена (аутокатализ). Пепсин в первую очередь гидролизует пептидные связи в белках, образованные ароматическими аминокислотами (фенилаланин, триптофан, тирозин) и несколько медленнее - образованные лейцином и дикарбоновыми аминокислотами. Пепсин - эндопептидаза, поэтому в результате его действия в желудке образуются более короткие пептиды, но не свободные аминокислоты.
|
|
|
|
|
Дата добавления: 2015-04-24; Просмотров: 1441; Нарушение авторских прав?; Мы поможем в написании вашей работы!