КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Острые миелоидные лейкозы (ОМЛ)
|
|
|
|
М0 - острый миелобластный лейкоз с минимальными признаками дифференцировки
М1 - острый миелобластный лейкоз без признаков вызревания М2 - острый миелобластный лейкоз с признаками вызревания М3 - гипергранулярный острый промиелоцитарный лейкоз М3v - микрогранулярный острый промиелоцитарный лейкоз М4 - острый миеломонобластный лейкоз М5а - острый монобластный лейкоз без признаков вызревания М5b - острый монобластный лейкоз с признаками вызревания М6 - острый эритробластный лейкоз (эритролейкоз) М7 - острый мегакариобластный лейкоз
2. Острые лимфобластные лейкозы (ОЛЛ) L1 - микролимфобластный ОЛЛ
L2 - ОЛЛ с типичными бластами L3 - макролимфобластный ОЛЛ
Таблица 14-13. Цитохимические особенности бластных клеток при острых лейкозах различного происхождения
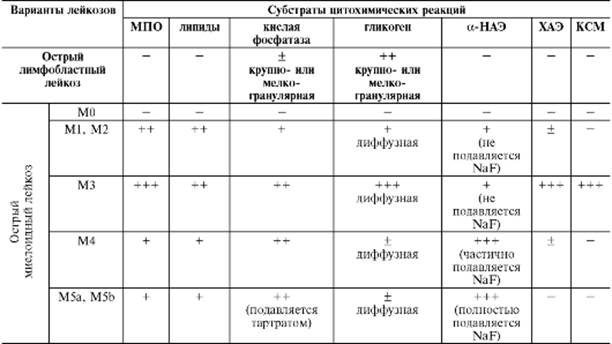

Примечание. МПО - миелопероксидаза; -НАЭ--нафтилацетатэстераза; ХАЭ - хлорацетатэстераза; КСМ – кислые сульфатированные мукополисахариды; «-» - отрицательная реакция; «+» - положительная реакция в единичных клетках; «+» - слабая реакция; «++» - умеренная реакция; «+++» - интенсивная реакция.
Выделенные нозологические формы острых лейкозов различаются по клиническим признакам и, что особенно важно, по ответу на цитостатическую медикаментозную терапию. У взрослых больных чаще встречаются миелобластный и лимфобластный, у детей - лимфобластный и (реже) недифференцированный варианты острого лейкоза.
Острый недифференцированный лейкоз, вариант М0. Морфологический субстрат опухоли представлен клетками II-III классов по современной схеме кроветворения, которые морфологически напоминают лимфобласты, но отличаются цитохимической интактностью (см. табл. 14-13). Из хромосомных аномалий для МО типичны моносомия 7, трисомия 4, 8, 13.
Острый миелобластный лейкоз. Представляет собой опухоль, исходящую из клетки-предшественницы миелопоэза и состоящую преимущественно из родоначальных клеток гранулоцитарного ряда - миелобластов. Выделяют 2 варианта ОМЛ - М1 и М2, для которых свойственны транслокации хромосом соответственно t(9;22) и t(8;21). В костном мозгу и крови у больных ОМЛ обнаруживаются многочисленные миелобласты средних размеров с узким ободком незернистой цитоплазмы и крупные миелобласты с обширной (иногда вакуолизированной) цитоплазмой с азурофильными гранулами и 1-2 палочками Ауэра (они могут определяться в более зрелых клетках гранулоцитарного ряда). Для зрелых нейтрофилов характерна гиперили гипосегментация ядер, «истощение» цитоплазматической зернистости.
Острый промиелоцитарный лейкоз. Для лейкозных клеток при этой форме острого лейкоза характерны транслокации хромосом: t(15;17), t(5;17), t(11;17). Атипичные промиелоциты содержат большое количество крупных (при гипергранулярном варианте - М3) или мелких (при микрогранулярном варианте - M3v) гранул и палочки Ауэра (нередко их 1О-2О и более). При слиянии палочек Ауэра образуются «faggot cells» - клетки с «пучком прутьев». Ядра атипичных промиелоцитов имеют рыхлую структуру хроматина, неправильную форму со смазанными контурами (иногда их форма неразличима из-за обилия гранул в цитоплазме).
Острый монобластный лейкоз. Существует два его варианта - без признаков вызревания клеток (М5а) и с признаками вызревания клеток (М5Ь), при которых обнаруживаются транслокационные перестройки хромосом t(9;11) и t(4;11). При М5а субстрат опухоли составляют монобласты крупных размеров с обширной
серо-голубой цитоплазмой, скудной азурофильной зернистостью и вакуолизацией. При М5b преобладающим типом клеток в костном мозгу и крови являются промоноциты (количество монобластов не превышает 10-15%).
Острый миеломонобластный лейкоз или вариант М4 острого лейкоза. В его основе лежат перестройки хромосомы 16: inv (16), t(16;16), t (8;16). Для М4 характерно присутствие в костном мозгу одновременно двух типов атипичных бластов - миелобластов и монобластов. В крови могут обнаруживаться также промоноциты.
Острый эритробластный лейкоз или М6. Наиболее частой цитогенетической аномалией при М6 является делеция длинного плеча хромосомы 5 или 7. Субстрат опухоли составляют клетки эритроидного ряда: эритронормобласты, гигантские, многоядерные эритрокариоциты с мегалобластическим оттенком, мегалобласты, аномальные эритроидные клетки с вакуолизированной и отростчатой цитоплазмой, с кариорексисом. В крови отмечаются выраженный анизо- и пойкилоцитоз, гиперхромия эритроцитов, появление в кровотоке эритроцитов с базофильной зернистостью, кольцами Кабо, эритробластов, полихроматофильных и оксифильных эритронормобластов.
Острый мегакариобластный лейкоз, вариант М7. Характерными цитогенетическими признаками М7 являются inv (3), t(3;3), t(1;22). В костном мозгу и крови выявляются мегакариобласты и их осколки. В крови обнаруживается тромбоцитоз (более 1000-109/л). Тромбоциты имеют гигантские размеры, могут склеиваться в «кучки», имеется качественный дефект клеток - «серые» тромбоциты (в результате дефекта α-гранул).
Острый лимфобластный лейкоз. Это опухоль, возникающая из клетки-предшественницы лимфопоэза. В костном мозгу при ОЛЛ выявляется тотальная лимфобластная метаплазия. Картина периферической крови при ОЛЛ характеризуется присутствием бластных клеток. Согласно ФАБ-классификации, в зависимости от цитоморфологических признаков лимфоидных клеток выделяют три цитологических варианта ОЛЛ: L1 (микролимфобластный), представленный однородной популяцией клеток малых размеров; L2 (с типичными бластами), характеризующийся неоднородностью клеток, среди которых преобладают крупные клетки, реже выявляются клетки средних и малых размеров; L3 (макролимфобластный), при котором обнаруживаются клетки только крупных размеров.
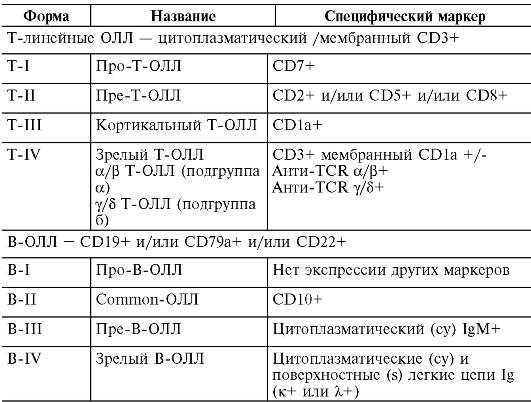
В 1995 г. Европейской группой по иммунологической характеристике лейкозов (European Group for the immunological characterization of leukemias, EGIL) была предложена классификация острых лимфобластных лейкозов по иммунологическому принципу. В соответствии с EGIL-классификацией их подразделяют на Т- и В-линейные лейкозы (табл. 14-14).
Таблица 14-14. EGIL-классификация острых лимфобластных лейкозов (ОЛЛ)
Хронические лейкозы. Несколько упрощенно классификацию хронических лейкозов можно представить в следующем виде. 1. Хронические миелопролиферативные лейкозы: Хронический миелолейкоз
• Ph-позитивный (типичный, взрослый тип)
• Ph-негативный (атипичный, ювенильный тип) Хронический миеломоноцитарный лейкоз Хронический нейтрофильный лейкоз Хронический эозинофильный лейкоз Хронический базофильный лейкоз
Хронический тучноклеточный лейкоз Истинная полицитемия (эритремия) Тромбоцитемия
Хронический идиопатический миелофиброз (сублейкемический миелоз)
2. Хронические лимфопролиферативные лейкозы:
В-клеточные хронические лимфолейкозы Хронический лимфолейкоз Волосатоклеточный лейкоз Парапротеинемические гемобластозы
• Множественная миелома (миеломная болезнь)
• Макроглобулинемия Вальденстрема
• Болезни тяжелых цепей
Т- и ЕК-клеточные (из ЕК-клеток - естественных киллеров) хронические лимфолейкозы
Болезнь Сезари (лимфоматоз кожи)
Лейкоз из больших гранулосодержащих лимфоцитов (ЕКклеток)
В отличие от острых лейкозов моноклоновая («доброкачественная») стадия хронических лейкозов является более продолжительной (годы, десятилетия). В развитии хронических лейкозов выделяют хроническую фазу, которая характеризуется длительным компенсированным течением, и фазу бластной трансформации, проявляющуюся бластным кризом с резким увеличением количества бластных клеток в костном мозгу и периферической крови (более 30%), прогрессированием анемии, тромбоцитопении и формированием внекостно-мозговых лейкемических инфильтратов.
Клинико-лабораторная картина хронических лейкозов характеризуются разнообразием признаков, строго специфичных для определенной формы заболевания. Наиболее распространенными вариантами хронических лейкозов являются хронический миелолейкоз, хронический миеломоноцитарный лейкоз, эритремия, сублейкемический миелоз, хронический лимфолейкоз, парапротеинемические гемобластозы и лимфоматоз кожи.
Хронический миелолейкоз. Одно из самых частых заболеваний в группе лейкозов. Морфологическим субстратом хронического миелолейкоза (ХМЛ) являются зрелые и созревающие клетки гранулоцитарного ростка кроветворения (лейкоэритробластическое отношение в костном мозгу увеличивается до 20:1 при 2:1-4:1 в норме). По современным представлениям ХМЛ возникает в результате соматической мутации в стволовой клетке-предшественнице миелопоэза, что в 85-95% случаев приводит к образованию хромосомного маркера - Ph'-хромосомы (см. рис. 14-12), являющейся продуктом делеции хромосомы 22 или транслокации дистальной части длинного плеча хромосомы 22 на хромосому 9. У детей ХМЛ встречается в двух вариантах - с филадельфийской хромосомой в кариотипе лейкозных клеток (взрослый тип заболевания) и без нее (ювенильный тип).
Течение хронического миелолейкоза как у взрослых, так и у детей разделяют на стадии или фазы заболевания в зависимости от степени зрелости клеточного состава крови. В хронической фазе типичного ХМЛ общее количество лейкоцитов в периферической крови колеблется от 50-109/л и более (у 25% больных - более 350-109/л). Отмечается нейтрофильный лейкоцитоз с выраженным ядерным сдвигом влево: обнаруживаются единичные миелобласты (2-3%), промиелоциты, миелоциты, метамиелоциты, палочко- и сегментно-ядерные формы гранулоцитов. Количество промиелоцитов и миелоцитов увеличивается по мере прогрессирования болезни при одновременном уменьшении числа палочкоядерных и сегментно-ядерных форм гранулоцитов. Отмечаются выраженная эозинофилия и базофилия (эозинофильно-базофильная ассоциация). В гранулоцитах обнаруживаются признаки дегенерации - псевдопельгеризация, низкая активность щелочной фосфатазы. У детей ювенильная (Ph-негативная) форма ХМЛ характеризуется высоким моноцитозом и тромбоцитопенией. В период бластной трансформации происходит резкое «омоложение» лейкоцитарной формулы за счет увеличения числа промиелоцитов и миелобластов (не менее 30%), прогрессирует цитопения (анемия, лейко-и тромбоцитопения), возникают лейкемические инфильтраты в коже, лимфоузлах, миокарде и других органах. При кариологическом исследовании выявляется поликлоновость патологических клеток (анэуплоидия), которая является основным признаком терминальной стадии - новым этапом опухолевой прогрессии.
Хронический миеломоноцитарный лейкоз характеризуется гиперклеточностью костного мозга за счет увеличения количества незрелых и зрелых клеток гранулоцитарного и моноцитарного ряда (промоноцитов, моноцитов). Миелобласты и монобласты составляют не более 5% от общего числа миелокариоцитов. В периферической крови обнаруживаются незрелые клетки нейтрофильного ряда (не более 10% от общего количества лейкоцитов), промоноциты, реже - бласты. В гранулоцитах - псевдопельгеризация, «истощение» зернистости в цитоплазме, отрицательная реакция на миелопероксидазу, низкая активность лейкоцитарной щелочной фосфатазы. В сыворотке крови и моче устанавливается высокое содержание лизоцима.
Эритремия (истинная полицитемия, болезнь Вакеза). Заболевание опухолевой природы, характеризующееся относительно доброкачественным течением. Источником роста опухоли является клетка-предшественница миелопоэза, основной субстрат опухоли - эритроциты. Наиболее характерны изменения со стороны периферической крови: количество эритроцитов достигает (6-12)-1012/л, уровень гемоглобина - 180-220 г/л, показатель гематокрита увеличивается до 60-80%. Уровень эритропоэтина в крови и моче в отличие от симптоматических эритроцитозов остается нормальным. Имеются лейко- и тромбоцитоз, уменьшается СОЭ, возрастает вязкость крови. Важным диагностическим признаком является увеличение массы циркулирующих эритроцитов, опосредующее гиперемию кожи и слизистых, окклюзию микрососудов и связанные с ней головные боли, боли в суставах, позвоночнике, эпигастрии и др.
Сублейкемический миелоз характеризуется гиперклеточностью костного мозга за счет гиперплазии гранулоцитарного, эритроидного и мегакариоцитарного ростков, а также миелофиброзом и миелосклерозом, при прогрессировании которых клеточность костного мозга постепенно снижается. В крови обнаруживаются незрелые клетки гранулоцитарного ряда, гипо- и гиперсегментированные нейтрофилы, миелобласты (1-5%). Количество тромбоцитов варьирует. Нередко выявляются крупные гипогранулярные тромбоциты:, аномальные мегакариоциты и их фрагменты. Гематологическими признаками сублейкемического миелоза, позволяющими дифференцировать его с ХМЛ, наряду с выраженным фиброзом и склерозом костного мозга, являются: увеличение общего количества лейкоцитов в крови не более 50-109/л (при ХМЛ более 50-109/л), умеренная базофилия при нормальном содержании эозинофилов (при ХМЛ сочетанное увеличение количества базофилов и эозинофилов - эозинофильно-базофильная ассоциация) и высокая активность щелочной фосфатазы в нейтрофилах (при ХМЛ активность лейкоцитарной щелочной фосфатазы низкая).
Хронический лимфолейкоз. Это опухоль иммунокомпетентной ткани, состоящая преимущественно из зрелых лимфоцитов, представленных в большинстве случаев В-клетками. Характерен лейкоцитоз; в мазках крови преобладают зрелые узкоцитоплазменные лимфоциты, содержание которых может доходить до 80% и более. Важным признаком служит появление дегенеративных форм лимфоцитов - теней Гумпрехта (результат раздавливания качественно неполноценных лимфоцитов при приготовлении гематологических мазков), голых ядер лимфоцитов и форм Ридера. Количество лимфоцитов в костном мозгу составляет не менее 30% всех миелокариоцитов. Разрастание лимфоидной ткани имеет место в лимфатических узлах, селезенке и печени, что сопровождается увеличением указанных органов.
Функциональная неполноценность образующих опухоль лимфоцитов приводит к нарушению иммунологического гомеостаза у больных, что, в свою очередь, становится причиной аутоиммунных конфликтов (аутоиммунные гемолитические анемии и тромбоцитопении); инфекционных осложнений (вследствие нарушения антителообразования) и т.д.
В отличие от хронического миелолейкоза бластные кризы наблюдаются крайне редко, не развивается также вторичной резистентности к цитостатическим препаратам.
Парапротеинемичские гемобластозы. К парапротеинемическим гемобластозам относятся миеломная болезнь (множественная миелома, плазмоцитома), макроглобулинемия Вальденстрема и болезни тяжелых цепей - опухоли В-клеточного происхождения. Главной особенностью данных лейкозов является сохранение способности В-клеток к дифференцировке до стадии иммуноглобулинсекретирующих клеток. При этом секретируемые ими иммуноглобулины отличаются однообразием структуры (моноклональные парапротеины), что объясняется их происхождением из одного клона опухолевых клеток. Парапротеины соответствуют разным вариантам нормальных иммуноглобулинов (чаще IgG или IgM), отличаясь от них строгой однотипностью тяжелых и легких цепей, либо представляют собой структурно аномальные молекулы иммуноглобулинов (изолированные фрагменты тяжелых цепей, свободные легкие цепи).
При множественной миеломе имеет место диссеминированная злокачественная пролиферация клона В-лимфоцитов на уровне плазматических клеток. Множественная миелома составляет около 1% всех злокачественных новообразований, частота встречаемости миеломной болезни колеблется в разных этнических группах
от 1 до 10 на 100 000 населения. Плазматические клетки наиболее часто пролиферируют диффузно по костному мозгу, но иногда формируют солитарную опухоль, называемую плазмоцитомой. Из-за остеолитических повреждений, обусловленных продукцией опухолевыми клетками остеокластактивирующих факторов (IL-1, TNF-α, TNF-β), развивается гиперкальциемия и связанные с ней поражения нервной, сердечно-сосудистой системы, почек, желудочно-кишечного тракта, формируются тромбоцитопения, анемия, лейкопения. Одновременно подавляется формирование нормальных плазматических клеток, что приводит к нарушению образования других иммуноглобулинов, развивается синдром возвратных инфекций.
В случае выявления парапротеинов при электрофорезе сыворотки крови обязательным является электрофоретическое исследование мочи. В 20% случаев миеломной болезни опухоль продуцирует только легкие цепи иммуноглобулинов, которые из-за низкой молекулярной массы быстро фильтруются в почках и могут не обнаруживаться в сыворотке, но выявляются в моче (протеинурия Бенс-Джонса). В связи с этим для подтверждения диагноза методом электрофореза определяют белок Бенс-Джонса в моче, который располагается недалеко от старта соответственно М-градиенту в сыворотке, между γ- и β-глобулинами.
Макроглобулинемия Вальденстрема характеризуется тем, что опухолевые В-лимфоциты продуцируют макромолекулярные моноклональные парапротеины класса IgМ. Из-за накопления высокомолекулярных белков характерны повышение вязкости крови, нарушение микроциркуляции, сладж-синдром, предрасположенность к тромбозам, геморрагический синдром. Болеют преимущественно мужчины после 60 лет. Картина крови характеризуется анемией, в патогенезе которой играют роль опухолевое подавление эритропоэза, кровопотери, нередко развивается лейкопения с нейтропенией, моноцитоз, тромбоцитопения присоединяется по мере прогрессирования заболевания. СОЭ всегда резко увеличена, в сыворотке гиперпротеинемия, а на электрофореграмме - М-градиент за счет IgМ. В моче белок Бенс-Джонса встречается примерно в 80% случаев, но количество его значительно меньше, чем при множественной миеломе. В отличие от миеломной болезни при макроглобулинемии Вальденстрема обычно обнаруживаются увеличение лимфоузлов и гепатоспленомегалия, но лимфоидная инфильтрация прогрессирует, как правило, относительно медленно.
При этом поражение костей и гиперкальциемия отмечаются редко. Проявления болезни исчезают при замещающей гемотрансфузии.
Болезни тяжелых цепей представляют собой В-клеточные лимфатические опухоли с разнообразной клинической и морфологической картиной и секрецией фрагментов тяжелых цепей различных классов иммуноглобулинов. Это обычно α-цепи, но могут быть также γ- или μ-цепи. Выделяют две формы течения болезни: абдоминальную и легочную. Абдоминальная форма характеризуется диффузной инфильтрацией слизистой тонкой кишки и мезентериальных лимфатических узлов лимфоидными и плазматическими клетками, макрофагами, тучными клетками. Поражение желудочно-кишечного тракта приводит к атрофии ворсинчатого эпителия и развитию синдрома мальабсорбции. Легочная форма протекает с бронхопульмональными поражениями и медиастинальной лимфаденопатией. Протеинурия отсутствует. Диагностика основана на иммунохимическом анализе сывороточных белков, позволяющем выявить тяжелые цепи иммуноглобулинов.
Болезнь Сезари (лимфоматоз кожи) проявляется генерализованной эритродермией, алопецией, поражением век, дистрофией ногтей, интенсивным зудом кожи. У 30% больных отмечается спленомегалия, у 60% - лимфаденопатия смешанной природы (одни лимфатические узлы увеличиваются реактивно вследствие присоединения кожных инфекций, другие - в связи с их лейкемической инфильтрацией). При исследовании трепанобиоптатов костного мозга обнаруживаются очаги лимфоидной инфильтрации из малых лимфоцитов и клеток Сезари - атипичных Т-лимфоцитов со светлой цитоплазмой и складчатыми (конволютными) ядрами. Содержание гемоглобина, количество эритроцитов и тромбоцитов в крови обычно сохраняется в пределах нормы. Характерен лимфоцитоз (до 70-109/л), наличие клеток Сезари в мазках крови.
|
|
|
|
|
Дата добавления: 2015-06-04; Просмотров: 3498; Нарушение авторских прав?; Мы поможем в написании вашей работы!