КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Ингибиторы
|
|
|
|
Метаболизм. Ферменты
Кинетика ферментативных реакций
Метаболизм. Ферменты
Кинетика ферментативной реакции (т. е. зависимость скорости реакции от ее условий) определяется в первую очередь свойствами катализатора, вследствие чего она значительно сложнее, чем кинетика некаталитических реакций (см. с. 28).
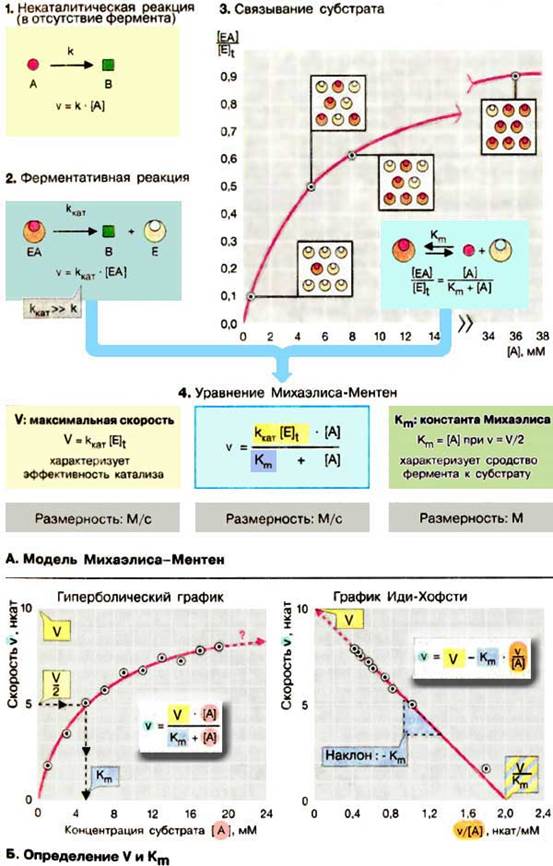
А. Модель Михаэлиса-Ментен
Многие соединения могут влиять на обмен веществ, модулируя активность соответствующих ферментов. Особенно важные функции при этом выполняют ингибиторы ферментов. Ингибиторами ферментов являются многие лекарственные вещества природного или синтетического происхождения (см. сс. 188, 250, 376 и 388). Метаболиты также могут быть ингибиторами ферментов в процессах регуляции (см. с. 116).
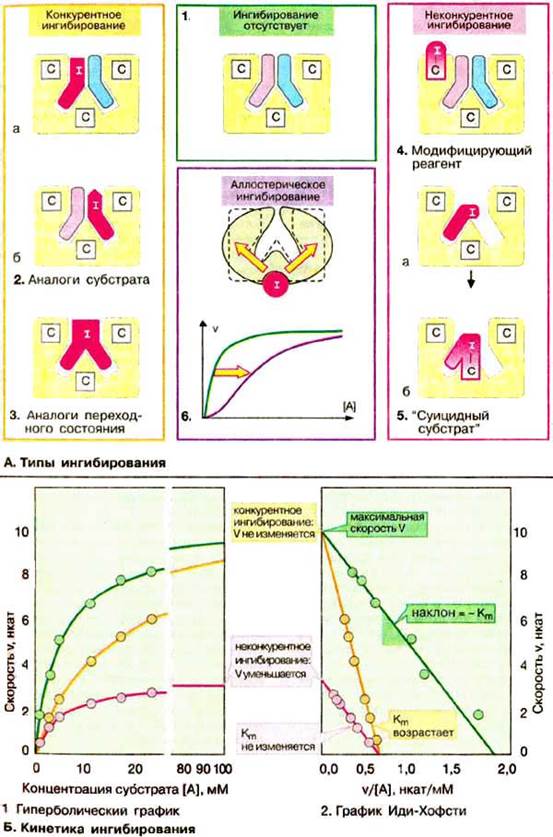
А. Типы ингибирования
Большинство ингибиторов ферментов действуют обратимо, т. е. не вносят в молекулу фермента каких-либо изменений после своей диссоциации. Однако существуют также необратимые ингибиторы ферментов, которые необратимо модифицируют целевой фермент. Принцип действия ингибитора, тип его ингибирования определяют путем сравнения кинетики реакции (см. с. 98) в присутствии ингибиторам без него (см. схему Б). Различают конкурентное (А, слева) и неконкурентное (А, справа) ингибирование. В регуляции обмена веществ важную роль играет аллостерическое ингибирование (А, 6).
Так называемые аналоги субстрата (2) имеют свойства, подобные свойствам субстрата целевого фермента. Они обратимо блокируют часть молекул имеющегося в наличии фермента, но не могут далее превращаться в продукт. Поэтому для достижения половины максимальной скорости реакции необходимы более высокие концентрации субстрата: в присутствии такого ингибитора константа Михаэлиса Km растет (Б). Субстрат в высоких концентрациях вытесняет ингибитор с фермента. Поэтому максимальная скорость V (см. с. 98) при этом типе торможения не претерпевает изменений. Так как субстрат и ингибитор конкурируют за место связывания на ферменте, данный тип торможения называют конкурентным. Аналоги переходного состояния (3) также действуют как конкурентные ингибиторы.
Если ингибитор реагирует с функционально важной группой фермента, не препятствуя связыванию субстрата, такое ингибирование называется неконкурентным (на схеме справа). В этом случае Km остается неизменной, напротив уменьшается концентрация функционально активного фермента [Е] t и, следовательно, максимальная скорость реакции V. Неконкурентные ингибиторы действуют как правило необратимо, поскольку они модифицируют функциональные группы целевого фермента (4).
В случае так называемых "суицидных субстратов" (5) речь идет о субстратных аналогах, содержащих дополнительно реакционную группу. Вначале они связываются обратимо, а затем образуют ковалентное соединение с активным центром фермента. Поэтому ингибирование такими соединениями проявляется как неконкурентное. Известным примером такого ингибитора является антибиотик пенициллин (см. с. 250).
Аллостерические ингибиторы связываются с отдельными участками фермента вне активного центра (6). Такое связывание влечет за собой конформационные изменения в молекуле фермента, которые приводят к уменьшению его активности (см. с. 118). Аллостерические эффекты встречаются практически только в случае олигомерных ферментов. Кинетику таких систем нельзя описать с помощью простой модели Михаэлиса-Ментен.
Б. Кинетика ингибирования
Конкурентное ингибирование легко можно отличить от неконкурентного при использовании графика Иди-Хофсти (см. с. 98). Как уже упоминалось, конкурентные ингибиторы влияют только на Km, но не на V. Полученные в отсутствие и в присутствии ингибитора прямые на графике пересекаются на оси ординат. Прямые для неконкурентного ингибирования имеют одинаковый наклон (Кm не изменяется), однако по мере увеличения концентрации ингибитора отрезки, отсекаемые этими прямыми на оси ординат, становятся все короче. Для аллостерических ферментов нельзя применять график Иди-Хофсти, имеющий в этом случае нелинейный характер (здесь не приведен).
Полный математический анализ ферментативной реакции приводит к сложным уравнениям, не пригодным для практического применения. Наиболее удобной оказалась простая модель, разработанная в 1913 г. Она объясняет характерную гиперболическую зависимость активности фермента от концентрации субстрата (1) и позволяет получать константы, которые количественно характеризуют эффективность фермента.
Модель Михаэлиса-Ментен исходит из того, что вначале субстрат А образует с ферментом E (З) комплекс, который превращается в продукт В намного быстрее, чем в отсутствие фермента. Константа скорости kкат (2) намного выше, чем константа некаталитической реакции k. Константу kкат называют еще «числом оборотов» поскольку она соответствует числу молекул субстрата, превращаемых в продукт одной молекулой фермента за 1 с. Согласно этой модели, активность фермента определяется долей комплекса EA от общей концентрации фермента [E] t, т. е., отношением [EA] / [E] t (З). С целью упрощения модель предполагает, что E, А и ЕА находятся в химическом равновесии согласно закону действующих масс (см. с. 24), что дает в итоге для диссоциации комплекса EA уравнение:
[E][A]/[EA] = Km Поскольку [E] t = [E] + [EA],
[EА] = [E] t [А]/(Кm + [А])
Из v = kкат[EA] (2) и предыдущего выражения получают уравнение Михаэлиса-Ментен (4).
Уравнение содержит две величины (два параметра), которые не зависят от концентрации субстрата [A], но характеризуют свойства фермента: это произведение kкат[E] t, соответствующее максимальной скорости реакции V при высокий концентрации субстрата, и константа Михаэлиса Кm, характеризующая сродство фермента к субстрату. Константа Михаэлиса численно равна той концентрации субстрата [A]. при которой ν достигает половины максимальной величины V (если v = V/2, то [A] / (Кm + [A]) = 1/2, т. е. Km = [А]). Высокое сродство фермента к субстрату характеризуется низкой величиной Кm и наоборот,
Модель Михаэлиса-Ментен основывается на нескольких не совсем реальных допущениях, таких, как необратимое превращение EA в E + В, достижение равновесия между E, A и EA, отсутствие в растворе других форм фермента, кроме E и EA. Только при соблюдении этих гипотетических условий Km соответствует константе диссоциации комплекса, а kкат — константе скорости peакции EA → E + В.
Б. Определение V и Кm
В принципе V и Кm можно определить по графику зависимости v от [A] (рис. слева). Так как v асимптотически достигает V с возрастанием концентрации субстрата [A], то затруднительно получить надежную величину V и Кm (рис. слева) путем экстраполяции.
Для удобства расчетов уравнение Михаэлиса-Ментен можно преобразовать так, чтобы экспериментальные точки лежали на прямой. При одном из таких графических преобразований в так называемом графике Иди-Хофсти (pиc. справа) строят график зависимости v от v/[A]. В этом случае точка пересечения прямой, полученной путем наилучшей линейной аппроксимации экспериментальных точек, с осью ординат соответствует V, а тангенс угла наклона равен -Km. Такой графический подход дня определения V и Кm также не оптимален. В настоящее время данные ферментативной кинетики обрабатывают быстрее и более объективно с помощью вычислительной техники.
Метаболизм. Ферменты
Лактатдегидрогеназа: структура
В этом разделе в качестве примера взаимосвязи структуры и функции фермента более подробно рассмотрена лактатдегидрогеназа [ЛДГ (LDH), КФ 1.1.1.27].
А. Лактатдегидрогеназа: структура
Активной формой лактатдегидрогеназы (молекулярная масса 144 кДа) является тетрамер из 4 субъединиц (1) Каждая субъединица образована пептидной цепью из 334 аминокислот (36 кДа). В тетрамере субъединицы занимают эквивалентные положения (1); каждый мономер содержит активный центр.
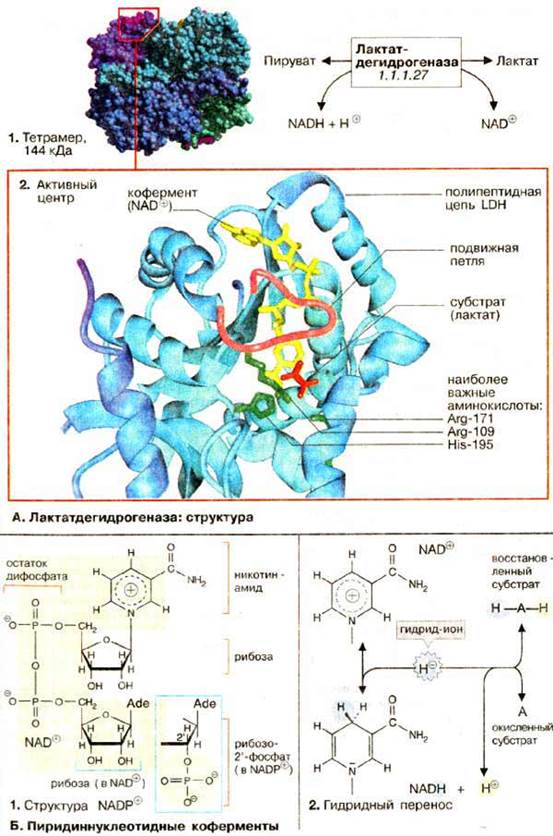
В организме млекопитающих имеются два различных типа субъединиц ЛДГ (H и M), незначительно различающиеся по аминокислотной последовательности; они могут ассоциировать в тетрамер случайным образом. Поэтому известно 5 различных изоферментов ЛДГ. В мышце сердца содержатся преимущественно тетрамеры, состоящие из Н-субъединиц (H от англ. heart), в ЛДГ печени и скелетных мышц преобладают М-мономеры.
Активный центр в субъединице ЛДГ схематически показан на рис. 2. При этом пептидный остов белка изображен в виде ленты (светло-голубой), дополнительно представлены молекулы: субстрата — лактата (красного цвета), кофермента НАД+ (желтого цвета) и три боковые цепи аминокислот (зеленого цвета), которые участвуют непосредственно в катализе. Кроме того, выделена пептидная петля (малинового цвета), образованная аминокислотными остатками 98-111. В отсутствие субстрата и кофермента эта структура раскрыта, что обеспечивает свободный доступ к субстратсвязывающему участку (не показано). На рисунке представлен блокированный активный центр в комплексе фермент-лактат-НАД+. Детали каталитического цикла лактатдегидрогеназы обсуждаются в следующем разделе.
Б. Пиридиннуклеотидные коферменты
Все дегидрогеназы нуждаются в коферменте для переноса восстановительных эквивалентов (см. с. 108). Наиболее широко распространены коферменты динуклеотидного типа (см. с. 86), в котором два нуклеозид-5'-монофосфата соединены фосфоангидридной связью. ЛДГ и многие другие дегидрогеназы нуждаются в никотинамидадениндинуклеотиде, сокращенно НАД+ (NAD+) (1). Обе нуклеотидных группы НАД+ построены из 5'-АМФ и нуклеотида, содержащего в качестве основания амид никотиновой кислоты (см. с. 354). Структурно (но не функционально) похожим коферментом является НАДФ+ (NADP+), в котором 2'-ОН-группы рибозы аденина дополнительно связаны с фосфатом. Несмотря на близкое структурное родство НАД+ и НАДФ+ осуществляют различные функции в обмене веществ (см. с. 114).
В окислительно-восстановительных реакциях пиридиннуклеотидного кофермента участвует только никотинамидное кольцо (2). Никотинамид является амидом пиридин-3-карбоновой (никотиновой) кислоты. В окисленной форме кольцо имеет ароматический характер и несет положительный заряд. По этой причине кофермент в окисленном состоянии обозначают как НАД+. При окислении лактата дегидрогеназа отщепляет от субстрата (AH2) два атома водорода [т. е. два электрона и два протона (2, середина)]. Однако на НАД+ переносится только гидрид-ион (H-, два электрона и один протон). Акцептором гидрид-иона является атом углерода в пара-положении к атому азота кольца НАД+. В этом месте образуется алифатическая СН2-группа, перестраиваются двойные связи кольца и исчезает положительный заряд (2, внизу). При окислении или восстановлении никотинамидного кольца изменяются также спектральные характеристики кофермента. Поэтому за реакцией можно легко следить фотометрически (см. с. 106).
Второй протон высвобождается в среду и, следовательно, правильное наименование восстановленной формы кофермента NADH + H+, а не NADH2.
Метаболизм. Ферменты
Лактатдегидрогеназа: механизм каталитической реакции
Механизм действия лактатадегидрогеназы (ЛДГ) можно представить на основе общих закономерностей ферментативного катализа, которые изложены на с. 96.
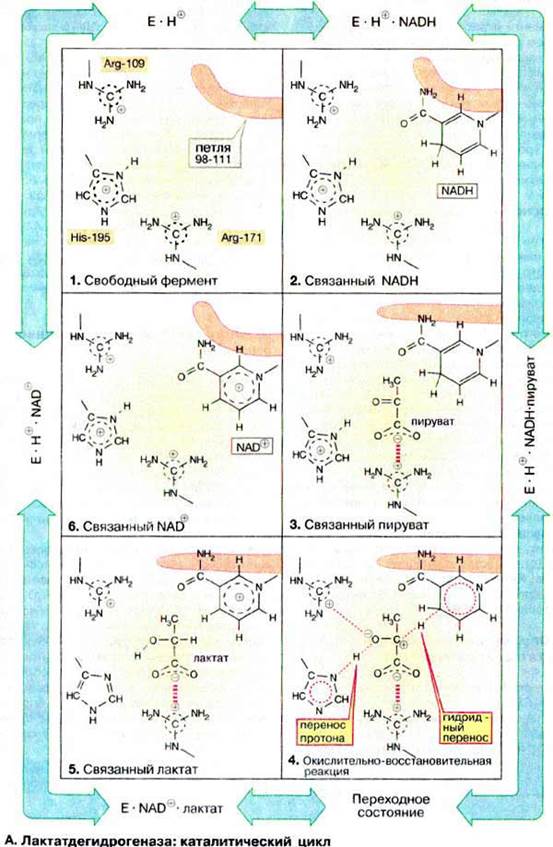
А. Лактатдегидрогеназа: каталитический цикл
ЛДГ катализирует передачу восстановительного эквивалента от лактата на НАД+ (NАD+) или от НАДН (NADH) на пируват.
L-лактат + НАД+ ↔ пируват + НАДН + H+
Равновесие реакции сдвинуто в сторону образования лактата (см. с. 24). Однако при высоких концентрациях лактата и НАД+ возможно окисление лактата в пируват. ЛДГ катализирует реакцию в обоих направлениях, но подобно всем ферментам не влияет на положение химического равновесия.
Из-за обратимости реакции каталитический процесс можно представить в виде движения по кругу. Каталитический цикл ЛДГ представлен на схеме в виде шести моментальных «снимков». Приведенные на схеме промежуточные стадии катализа очень кратковременны и поэтому строго не доказаны. Однако их существование подтверждается множеством экспериментальных данных
В активном центре ЛДГ принимают участие многие аминокислотные остатки. Они способствуют присоединению субстратов и кофермента или непосредственно участвуют в одной из стадий катализа. Здесь показаны лишь три особенно важные боковые цепи аминокислот: положительно заряженная гуанидиновая группа аргинина-171 связывает карбоксильную группу субстрата с помощью электростатического взаимодействия, имидазольная группа гистидина-195 принимает участие в кислотно-основном катализе, боковая цепь аргинина-109 важна для стабилизации переходного состояния. В противоположность His-195, меняющему свой заряд во время катализа, оба упомянутых остатка аргинина протонированы постоянно. Кроме этих трех остатков важную роль играет пептидная петля 98-111, показанная схематически (окрашена в малиновый цвет) на рис. 103. Ее функция состоит в том, чтобы после связывания субстрата и кофермента закрыть активный центр и исключить доступ молекул воды во время переноса электронов.
Рассмотрим теперь отдельные стадии катализируемого ЛДГ восстановления пирувата: в свободном ферменте (1) His-195 протонирован, в связи с чем эта форма обозначена как E ∙ H+. Кофермент НАДН связывается первым (2), а за ним - пируват (3) Важно, что в молекуле фермента карбонильная группа пирувата и никотинамидное кольцо кофермента в активированном состоянии расположены друг относительно друга оптимально и такая ориентация фиксирована (сближение и ориентирование субстратов). Затем закрывается петля 98-111 над активным центром. Сильно пониженная полярность в области активного центра облегчает образование переходного состояния (4; доступ воды закрыт). В переходном состоянии гидрид-ион H- (см. с. 104) переносится с кофермента на карбонильный углерод (перенос группы). При этом временно образующийся энергетически невыгодный отрицательный заряд на кислороде стабилизируется электростатическим взаимодействием с Arg-109 (стабилизация переходного состояния). Одновременно осуществляется перенос протона с Нis-195 на атом кислорода (перенос группы), приводя к образованию связанных с ферментом лактата и НАД+ (5). После открытия петли лактат диссоциирует с фермента и временно незаряженная имидазольная группа Нis-195 снова присоединяет протон из окружающей воды (6). Наконец, освобождается также окисленный кофермент НАД+ и снова достигается исходное состояние (1). При окислении лактата в пируват протекают то же стадии, но в противоположном направлении
Входящий в уравнение реакции протон присоединяется не одновременно с NADH а после освобождения лактата, т. е. между стадиями (5) и (6) предшествующего цикла.
Метаболизм. Ферменты
Ферментативный анализ
Ферменты играют важную роль в биохимическом анализе. В биологических материалах, например в жидкостях организма, с помощью определения каталитической активности можно обнаружить ферменты в ничтожно малых концентрациях. Ферменты можно использовать как реагенты для определения концентраций метаболитов, например уровня глюкозы в крови (схема В). В большинстве ферментативных анализов применяется фотометрия.
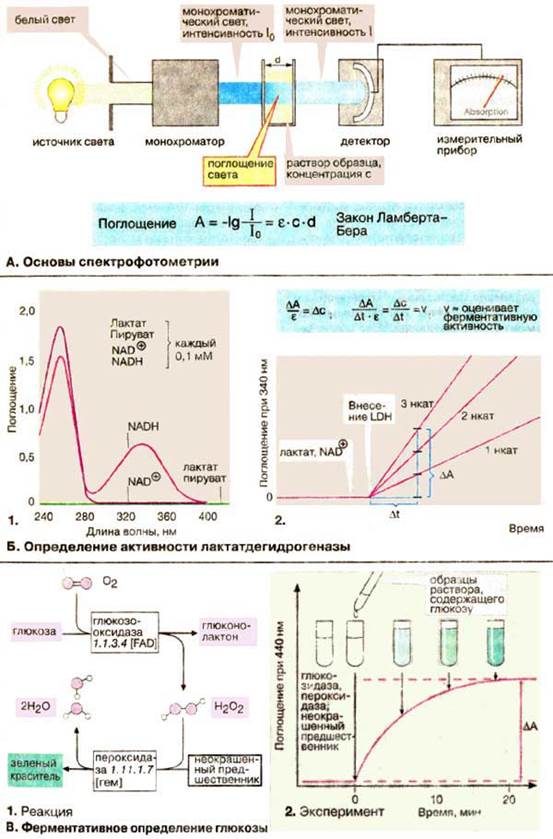
А. Основы спектрофотометрии
Многие молекулы поглощают свет в видимой или ультрафиолетовой области спектра. Это свойство можно использовать для определения концентраций. Величина поглощения зависит от типа и концентрации вещества, а также от длины волны используемого света. Поэтому применяют монохроматический свет, т. е. свет определенной длины волны, который можно выделить из белого света с помощью монохроматора. Монохроматический свет интенсивности I0 проходит через прямоугольную ячейку из стекла или кварца (кювету), в которой находится раствор поглощающего вещества. Интенсивность I выходящего света, ослабленного поглощением, измеряется с помощью детектора. Поглощение света (А) раствора (оптическая плотность) определяется как отрицательный логарифм отношения I / I0. Закон Ламберта-Бера гласит, что А пропорциональна концентрации (с) вещества и толщине (d) слоя раствора. Коэффициент экстинкции ε зависит, как было указано выше, от типа вещества и длины волны.
Б. Определение активности лактатдегидрогеназы
Определение активности лактатдегидрогеназы [ЛДГ (LDH)] основано на том факте, что восстановленный кофермент НАДН + H+ поглощает свет при 340 нм, в то время как у НАД+ (NAD+) при этой длине волны поглощение отсутствует. Спектры поглощения (т. е. графики зависимости А от длины волны) субстрата и кофермента в ЛДГ-реакции показаны на рис. Б1.
Различия в поглощении НАД+ и НАДН между 300 и 400 нм обусловлены изменениями никотинамидного кольца при окислении или восстановлении (см. с. 102). Для определения активности в кювету помещают прежде всего растворы лактата и НАД+ и регистрируют поглощение при постоянной длине волны 340 нм. Некаталитическая реакция протекает с очень низкой скоростью. Поэтому измеряемые количества НАДН образуются только после добавления ЛДГ. Так как скорость увеличения поглощения ΔA/Δt по закону Ламберта-Бера пропорциональна скорости реакции Δc/Δt, активность ЛДГ можно рассчитать с помощью коэффициента экстинкции ε при 340 нм или путем сравнения со стандартным раствором.
В. Ферментативное определение глюкозы
Большинство биомолекул не поглощают свет в видимой или ультрафиолетовой областях спектра. Кроме того, они обычно присутствуют в смеси с другими соединениями, которые также дают аналогичные химические реакции. Обе трудности можно преодолеть с помощью подходящего фермента для избирательного превращения определяемого метаболита в окрашенное вещество, которое далее определяют по интенсивности поглощения света.
Обычный метод определения глюкозы в крови (см. с. 162) основан на двух последовательных реакциях:
1) образование глюконолактона и пероксида водорода H2O2 под действием фермента глюкозооксидазы;
2) окисление бесцветного вещества пероксидом водорода в окрашенное зеленое соединение в реакции, катализируемой пероксидазой.
Когда вся имеющаяся в пробе глюкоза израсходована, количество образованного окрашенного вещества можно определить по светопоглощению, которое прямо пропорционально первоначальному содержанию глюкозы.
Метаболизм. Ферменты
Ферментативный анализ
Ферменты играют важную роль в биохимическом анализе. В биологических материалах, например в жидкостях организма, с помощью определения каталитической активности можно обнаружить ферменты в ничтожно малых концентрациях. Ферменты можно использовать как реагенты для определения концентраций метаболитов, например уровня глюкозы в крови (схема В). В большинстве ферментативных анализов применяется фотометрия.
А. Основы спектрофотометрии
Многие молекулы поглощают свет в видимой или ультрафиолетовой области спектра. Это свойство можно использовать для определения концентраций. Величина поглощения зависит от типа и концентрации вещества, а также от длины волны используемого света. Поэтому применяют монохроматический свет, т. е. свет определенной длины волны, который можно выделить из белого света с помощью монохроматора. Монохроматический свет интенсивности I0 проходит через прямоугольную ячейку из стекла или кварца (кювету), в которой находится раствор поглощающего вещества. Интенсивность I выходящего света, ослабленного поглощением, измеряется с помощью детектора. Поглощение света (А) раствора (оптическая плотность) определяется как отрицательный логарифм отношения I / I0. Закон Ламберта-Бера гласит, что А пропорциональна концентрации (с) вещества и толщине (d) слоя раствора. Коэффициент экстинкции ε зависит, как было указано выше, от типа вещества и длины волны.
Б. Определение активности лактатдегидрогеназы
Определение активности лактатдегидрогеназы [ЛДГ (LDH)] основано на том факте, что восстановленный кофермент НАДН + H+ поглощает свет при 340 нм, в то время как у НАД+ (NAD+) при этой длине волны поглощение отсутствует. Спектры поглощения (т. е. графики зависимости А от длины волны) субстрата и кофермента в ЛДГ-реакции показаны на рис. Б1.
Различия в поглощении НАД+ и НАДН между 300 и 400 нм обусловлены изменениями никотинамидного кольца при окислении или восстановлении (см. с. 102). Для определения активности в кювету помещают прежде всего растворы лактата и НАД+ и регистрируют поглощение при постоянной длине волны 340 нм. Некаталитическая реакция протекает с очень низкой скоростью. Поэтому измеряемые количества НАДН образуются только после добавления ЛДГ. Так как скорость увеличения поглощения ΔA/Δt по закону Ламберта-Бера пропорциональна скорости реакции Δc/Δt, активность ЛДГ можно рассчитать с помощью коэффициента экстинкции ε при 340 нм или путем сравнения со стандартным раствором.
В. Ферментативное определение глюкозы
Большинство биомолекул не поглощают свет в видимой или ультрафиолетовой областях спектра. Кроме того, они обычно присутствуют в смеси с другими соединениями, которые также дают аналогичные химические реакции. Обе трудности можно преодолеть с помощью подходящего фермента для избирательного превращения определяемого метаболита в окрашенное вещество, которое далее определяют по интенсивности поглощения света.
Обычный метод определения глюкозы в крови (см. с. 162) основан на двух последовательных реакциях:
1) образование глюконолактона и пероксида водорода H2O2 под действием фермента глюкозооксидазы;
2) окисление бесцветного вещества пероксидом водорода в окрашенное зеленое соединение в реакции, катализируемой пероксидазой.
Когда вся имеющаяся в пробе глюкоза израсходована, количество образованного окрашенного вещества можно определить по светопоглощению, которое прямо пропорционально первоначальному содержанию глюкозы.
Метаболизм. Ферменты
Окислительно-восстановительные коферменты
А. Коферменты: функции
Многие ферментативные реакции включают перенос электронов или групп атомов с одного субстрата на другой. В таких реакциях всегда принимают участие вспомогательные соединения (коферменты), которые выполняют функцию промежуточных переносчиков атомов или функциональных групп. Так как эти вещества каталитически не активны, правильнее было бы их называть косубстратами. Ферменты обычно высокоспецифичны к своим субстратам (см. с. 94), коферменты же взаимодействуют со многими ферментами, обладающими различной субстратной специфичностью.
По способам взаимодействия с ферментом различают растворимые коферменты и простатические группы. Растворимый кофермент (1) присоединяется во время реакции к молекуле фермента подобно субстрату, химически изменяется и затем снова освобождается. Первоначальная форма кофермента регенерируется во второй, независимой реакции. Простетической группой (2) называется кофермент, который прочно связан с ферментом и во время реакции его не покидает. Группа, связавшаяся с коферментом, далее переносится на следующий субстрат или другую молекулу кофермента (на схеме 2 не показано).
Б. Окислительно-восстановительные коферменты
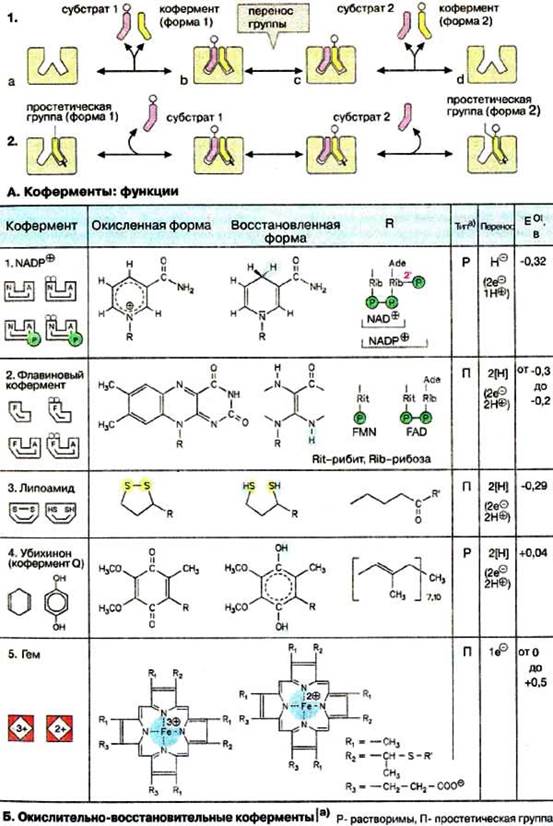
Все оксидоредуктазы (см. с. 94) нуждаются в коферменте. Наиболее важные коферменты представлены на схеме. Они могут действовать в растворимой форме (Р) или в виде простетической группы (П). Окислительно-восстановительные реакции, наряду с переносом электронов, часто включают перенос одного или двух протонов. Поэтому обычно принято говорить о переносе восстановительных эквивалентов. Стандартный потенциал Eo' простетической группы (см. с. 24) может значительно отличаться в зависимости от окружения в молекуле фермента.
Пиридиннуклеотиды НАД+ (NAD+) и НАДФ+ (NADP+) (1) широко распространены как коферменты дегидрогеназ. Они переносят гидрид-ион (2е- и 1 H+, см. с. 102) и действуют всегда в растворимой форме. НАД+ передает восстановительный эквивалент из катаболического пути вдыхательную цепь и тем самым участвует в энергетическом обмене. HАДФ+, напротив, является самым важным восстановителем при биосинтезе (см. с. 114).
Флавиновые коферменты ФМН (FMN) и ФАД (FAD) (2, см. с. 86) найдены в дегидрогеназах, оксидазах и монооксигеназах. Обычно оба соединения ковалентно связаны с ферментами. Активной группой обоих коферментов является флавин (изоаллоксазин), имеющий сопряженную систему из трех колец, которая может при восстановлении принимать два электрона и два протона. В ФМН к флавину присоединен фосфорилированный полиол рибит. ФАД состоит из ФМН, связанного с АМФ. Оба соединения являются функционально близкими коферментами.
В липоевой кислоте (3) функцию окислительно-восстановительного центра выполняет внутримолекулярный дисульфидный мостик. Активная липоевая кислота ковалентно связана с остатком лизина (R') молекулы фермента. Липоевая кислота прежде всего участвует в окислительном декарбоксилировании 2-кетокислот (см. с. 136). Дисульфидный мостик также содержится в пептидном коферменте глутатионе (см. с. 278).
Функция убихинона (кофермента Q, 4) как переносчика восстановительного эквивалента в дыхательной цепи будет рассмотрена на с. 142. При восстановлении хинон превращается в ароматический гидрохинон (убихинол). Похожие системы хинон/гидрохинон принимают участие в реакциях фотосинтеза (см. с. 132). К этому классу окислительно-восстановительных систем принадлежат также витамины Е и К (см. с. 352).
Группа гема (5) является окислительно-восстановительным кофактором в дыхательной цепи (см. с. 144), фотосинтезе (см. с. 130), а также в монооксигеназах (см. с. 310) и пероксидазах. В отличие от гемоглобина в этих случаях ион железа меняет валентность. На рисунке показан гем в цитохроме с, ковалентно связанный с двумя остатками цистеина (R2) белка.
Метаболизм. Ферменты
Коферменты переноса групп
В данном разделе рассматриваются коферменты, участвующие в реакции переноса групп. Кобамид (кофермент В12) будет рассмотрен на с. 356.
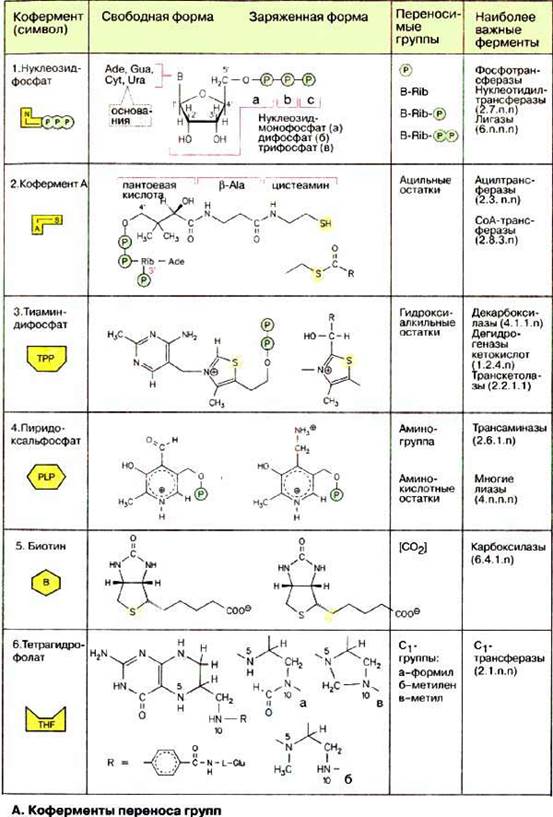
А. Коферменты переноса групп
Нуклеозидфосфаты (1) являются не только исходными соединениями в биосинтезе нуклеиновых кислот, они обладают также функциями коферментов, служат для запасания энергии и участвуют в цепи переноса энергии (см. с. 196) в эндоэргических процессах. Метаболические интермедиаты часто становятся реакционноспособными («активированными») при присоединении фосфат содержащих остатков (фосфорилирование). Так, присоединение нуклеозиддифосфатных остатков делает реакционноспособными исходные соединения в синтезе полисахаридов и липидов (см. с. 112). Лигазы катализируют сшивание соединений за счет энергии нуклеозидтрифосфатов.
Остатки жирных кислот активируются путем переноса на кофермент А (2). В коферменте А пантетеин через фосфоангидридную связь присоединен к 3'-фосфо-АДФ. Пантетеин состоит из трех компонентов, связанных амидными связями: пантоевой кислоты, β-аланина и цистеамина, т. е. двух биогенных аминов, образованных путем декарбоксилирования соответственно аспартата и цистеина (см. с. 182). Пантотеновая кислота, образованная из пантоевой кислоты и β-аланина, в организме человека играет роль витамина (см. с. 354). При реакции тиоловой группы остатка цистеамина с карбоновой кислотой образуется тиолсложноэфирная связь, как, например, в ацетил-КоА (ацетил-СоА). Эта реакция высоко эндоэргична и поэтому сопряжена с экзоэргическими процессами. Тиоэфир, каким является ацил-КоА, представляет собой активированную форму карбоновой кислоты, так как образующий ее ацильный остаток может легко переноситься на другую молекулу. Этот принцип часто используется при метаболических превращениях.
Тиаминдифосфат (TPP, 3) активирует альдегиды и кетоны и переносит их в виде гидроксиалкильных групп на другую молекулу. Этот способ переноса важен, например, в транскетолазной реакции (см. с. 154). Гидроксиалкильные остатки участвуют также в декарбоксилировании кетокислот. Они либо высвобождаются в виде альдегидов, либо переносятся на липоамидные остатки, как в случае дегидрогеназ 2-кетокислот (см. с. 128).
Пиридоксальфосфат (PLP) (4) — наиболее важный кофермент в метаболизме аминокислот, его роль при трансаминировании будет подробно рассмотрена на с. 180. Пиридоксальфосфат принимает участие и в других реакциях аминокислот, таких, как декарбоксилирование и дегидратирование. Представленная здесь альдегидная форма в свободном виде не встречается. В отсутствие субстрата альдегидная группа связана с аминогруппой лизинового остатка фермента в виде альдимина («шиффово основание»).
Карбоксилазы содержат в качестве кофермента биотин (5). Он связан амидной связью с боковой цепью лизинового остатка фермента. Биотин реагирует с гидрокарбонатом (НСО3-) в присутствии АТФ с образованием N-карбоксибиотина. Эта активированная форма диоксида углерода может быть перенесена на другую молекулу. Примерами биотинзависимых реакций являются образование оксалоацетата из пирувата (см. с. 156) и синтез малонил-КоА из ацетил-КоА (см. с. 170).
Тетрагидрофолат [ТГФ (THF), 6] является коферментом, который может переносить C1-остатки в различных состояниях окисления. ТГФ образуется из витамина фолиевой кислоты (см. с. 354) двойным гидрированием птеринового кольца. C1-фрагменты присоединяются к N-5, N-10 или к обоим атомам азота. Наиболее важными производными тетрагидрофолата являются:
а) N10-формил-ТГФ, в котором C1-остаток находится в виде карбоксильной группы,
б) N5, N10-метилен-ТГФ, в котором C1-остаток находится в виде альдегида и
в) N5-метил-THF, где C1 находится в виде спирта.
Переносимый ТГФ C1-фрагмент играет важную роль, например, в синтезе пуриновых нуклеотидов (см. с. 190), дезокситимидинмонофосфата (см. с. 192) и метионина (см. с. 406).
|
|
|
|
Дата добавления: 2015-07-13; Просмотров: 1686; Нарушение авторских прав?; Мы поможем в написании вашей работы!