КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Плазмоцитома: клинические проявления, диагностика, лечение
Парапротеинемические лейкозы – опухоли, секретирующие моноклональные Ig или их фрагменты (множественная миелома, солитарная плазмоцитома, макроглобулинемия Вальденстрема, лимфомы с моноклональной секрецией Ig, болезни тяжелых Ig, трудноклассифицируемые Ig-секретирующие опухоли).
Общие клинические особенности парапротеинемических лейкозов: синдром белковой патологии; нефропатия (вторичный амилоидоз); полинейропатия; гипервискозность крови; нарушения гемостаза; нарушения гуморального иммунитета; гиперурикемический синдром (типа вторичной подагры).
Плазмоцитома (множественная миелома, болезнь Рустицкого-Калера) – злокачественная опухоль из плазматических клеток (В-лимфоцитов конечной стадии дифференцировки), секретирующих парапротеины (в 70% - патологический моноклональный IgG, в 20% - патологический моноклональный IgA, в 5% - легкие цепи иммуноглобулинов).
Этиология плазмоцитомы достоверно неизвестна, развитию заболевания способствуют следующие факторы:
а) генетическая предрасположенность
б) дефекты Т-клеточной супрессии
в) влияние хронической антигенной стимуляции
г) повреждения генома (в результате действия радиации, химических веществ, ЛС, вирусов и др.)
Патогенез: опухолевая трансформация на уровне В-лимфоцитов-клеток памяти или плазмобластов, сохраняющих способность созревать и дифференцироваться в плазматические клетки ® формирование клона плазматических клеток, продуцирующих однородные по иммунологическим признакам Ig (парапротеины, М-протеины) или легкие цепи иммуноглобулинов (белки Бенс-Джонса) ®
1) хроническая стадия – миеломные клетки с низкой пролиферативной активностью, не выходящие за пределы костного мозга и кортикального слоя кости ® онкогенные мутации в опухолевом клоне ®
2) острая стадия (терминальная) – миеломные клетки с высокой пролиферативной активностью, выходящие за пределы костного мозга и метастазирующие во внутренние органы, приводящие к тяжелой миелодепрессии, выраженной интоксикации и др. проявлениям.
Клинические проявления плазмоцитомы:
1) бессимптомный период (5-15 лет) – общее состояние удовлетворительное, клиничеких проявлений поражения внутренних органов и костной системы нет; в ОАК - высокое СОЭ, при электрофорезе белков сыворотки крови – М-компонент, в ОАМ – необъяснимая протеинурия
2) развернутая клиническая стадия – жалобы на выраженную общую слабость, снижение трудоспособности, головокружение, снижение аппетита, похудание и др.; характерны ряд клинических синдромов:
а) синдром костной патологии – миеломатозные опухоли различных размеров в ребрах, грудине, позвоночнике, черепе, конечностях, проявляющиеся болями (вначале летучими, кратковременными, затем постоянными, интенсивными, продолжительными); остеопороз и литические повреждения костей из-за усиления их резорбции; частые патологические переломы (особенно позвоночника с компрессией позвонков)
б) синдром повышенной вязкости крови – нарушение микроциркуляции из-за гиперпротеинемии: неврологические нарушения (головная боль, головокружение, пошатывание при ходьбе, ощущение онемения и слабости в руках и ногах); нарушения периферического кровотока в конечностях (вплоть до гангрены); нарушение зрения (снижение остроты, мелькание мушке и пятен перед глазами); геморрагический синдром (кровоточивость слизистых, кожные геморрагии – из-за «окутывания» тромбоцитов парапротеинами с нарушением их агрегации).
в) поражение системы кроветворения – анемический синдром (из-за сокращения красного ростка костного мозга при выраженной миеломной пролиферации), в тяжелых случаях – нейтропения и тромбоцитопения.
г) синдром белковой патологии – обусловлен гиперпродукцией парапротеинов (патологических Ig или легких цепей иммуноглобулинов – белка Бенс-Джонса); гиперпротеинемия (общий белок может достигать 150-180 г/л), диспротеинемия (гипоальбуминемия, гипергаммаглобулинемия); М-компонент на электрофореграмме белков сыворотки крови; стойкая протеинурия; амилоидоз (AL-типа)
д) синдром поражения почек (миеломная нефропатия) – парапротеины накапливаются в почечных канальцах и реабсорбируются их клетками, вызывая повреждения; характерны протеинурия, гематурия, отеки, признаки ХПН
е) синдром висцеральной патологии – обусловлен развитием экстрамедуллярных поражений (чаще печени, селезенки, реже – плевральных оболочек, ЖКТ)
ж) синдром вторичного иммунодефицита – обусловлен снижением уровня нормальных иммуноглобулинов сыворотки, нарушением антителообразования, уменьшением гранулоцитов и их функциональной активности; проявляется предрасположенностью к инфекциям (частые ОРВИ, опоясывающий герпес и др.)
з) неврологический синдром – обусловлен плазмоклеточной инфильтрацией твердой мозговой оболочки, экстрадуральными миеломами, изменениями костей черепа и позвонков, компрессией нервных стволов миеломными разрастаниями; клинически – периферическая нейропатия (мышечная слабость, снижение поверхностной чувствительности, парестезии и др.), сдавление корешков спинного мозга, поражение черепно-мозговых нервов
к) гиперкальциемический синдром – обусловлен вымыванием кальция из костей в связи с их резорбцией; клинически – тошнота, рвота, сонливость, нарушения сознания, дезориентированность (если уровень кальция более 3,5 ммоль/л – необходимы прием внутрь более 3 л минеральной воды + форсированный диурез).
3) терминальная стадия – тяжелое клиническое течение, резкое обострение всей симптоматики, прогрессирование ХПН, анемии, тяжелых инфекционныз процессов, быстрое разрушение костей с прорастанием миеломой окружающих мягких тканей, внутренних органов и гибелью больного.
Диагностика плазмоцитомы:
1) ОАК: очень высокая СОЭ, могут быть анемия, лейкопения, тромбоцитопения
2) БАК: увеличение общего белка за счет гипергаммаглобулинемии; гиперкальциемия; повышение концентрации креатинина и мочевины
3) ОАМ: протеинурия, выявление белка Бенс-Джонса
3) иммунохимическое исследование крови и мочи: М-градиент во фракции гамма-глобулинов
4) миелограмма – плазмоцитоз (более 10% плазматических клеток в биоптате); плазматические клетки имеют широкую интенсивно синюю цитоплазму с четкими граница и овальное экцентрично расположенное ядро с колесовидным рисунком хроматина
5) рентгенологическое исследование костей – зоны лизиса костной ткани; компрессионные поражения позвоночника; «штампованные» литические поражения костей черепа и др.; различают 3 рентгенологические формы плазмоцитомы – остеолитическую (зоны просветления различной величины); кистозно-трабекулярную («мыльные пузыри»); диффузный остеопороз.
Лечение:
1. Основной метод – химиотерапия: мелфалан 9 мг/м2 + преднизолон 100-200 мг (режим М + Р) – стандарт терапии первой линии, 6-12 курсов --> ремиссия --> поддерживающая терапия интерфероном-α в дозе 3 млн. ЕД/м2 3 раза в неделю. При плазмоцитоме рецидивы неизбежны; другие алкилирующие препараты: алкеран, сарколизин, циклофосфан, препараты нитрозомочевины и т.д.
2. Лучевая терапия паллиативна, применяется при устойчивости плазмоцитомы к цитостатикам.
3. На фоне полихимиотерапии возможна трансплантация костного мозга
4. Симптоматическое лечение: остеопороз – бифосфонаты, гиперкальциемия – гидратация с форсированным диурезом; для снижения гипервязкости – плазмаферез; анемия – гемотрансфузии; инфекции – АБ и др.
62. Геморрагические диатезы: этиология, патогенез, классификация. Клиника, диагностика и лечение тромбоцитопенической пурпуры.
Геморрагические диатезы – патологические состояния, характеризующиеся повышенной кровоточивостью в результате недостаточности одного или нескольких элементов гемостаза.
Этиопатогенетическая классификация геморрагических диатезов:
а) наследственные – связаны с генетически детерминированными патологическими изменениями сосудистой стенки, аномалиями мегакариоцитов, тромбоцитов, адгезивных белков плазмы и плазменных факторов свертывающей системы крови.
б) приобретенные – обусловлены следующими причинами:
1) вследствие первичного поражения сосудистой стенки: наследственная геморрагическая телеангиэктазия Рандю-Ослера; геморрагический васкулит Шенлейна-Геноха; синдром Элерса-Данло, гиповитаминозы С и В и др.
2) вследствие первичного поражения мегакариоцитарно-тромбоцитарного ростка:
1. тромбоцитопении (идиопатическая тромбоцитопеническая пурпура, повышенное потребление тромбоцитов при ДВС, перераспределение тромбоцитов и их депонирование в селезенке)
2. тромбоцитопатии (тромбастения Глянцмана, болезнь фон Виллебранда)
3) вследствие нарушения свертывания крови (коагулопатии): витамин К-зависимые (при недостаточности функции печени, нарушении всасывания витамина К, алиментарной недостаточности витамина К и др.); печеночная недостаточность с дефицитом факторов свертывания и др.; патологические ингибиторы свертывания («волчаночный антикоагулянт»)
4) вследствие комплексных нарушений различных звеньев свертывающей системы: острые синдромы диссеминированного внутрисосудистого свертывания
Типы кровоточивости:
1) капиллярный (микроциркуляторный, петехиально-пятнистый, синячковый) – петехиальные высыпания, синяки и экхимозы на коже и слизистых; часто сочетается с кровоточивостью слизистых - носовыми кровотечениями, меноррагиями (тромбоцитопении, тромбоцитопатии)
2) гематомный – болезненные, напряженные кровоизлияния в подкожную клетчатку, мышцы, крупные суставы, брюшину и забрюшинное пространство; иногда почечные и желудочно-кишечные кровотечения (гемофилия А и В)
3) смешанный капиллярно-гематомный (синячково-гематомный) – петехиально-синячковые высыпания в сочетании с обширными плотными кровоизлияниями и гематомами; кровоизлияния в суставы не характерны
4) васкулитно-пурпурный – геморрагические или эритематозные (на воспалительной основе) высыпания различной величины; легко возникают в местах сдавления кожи поясом, носками (васкулиты)
5) ангиоматозный – упорные, строго локализованные и привязанные к локальной сосудистой патологии кровотечения (телеангиэктазии, гематомы)
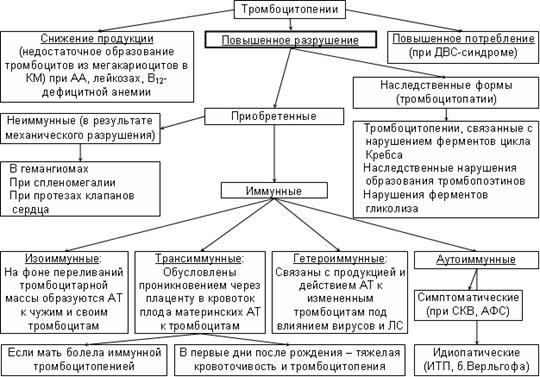 Тромбоцитопеническая пурпура – группа заболеваний, объединяемых по принципу единого патогенеза тромбоцитопении (укорочение продолжительности жизни тромбоцитов, обусловленное действием АТ к ним или другими механизмами их разрушения).
Тромбоцитопеническая пурпура – группа заболеваний, объединяемых по принципу единого патогенеза тромбоцитопении (укорочение продолжительности жизни тромбоцитов, обусловленное действием АТ к ним или другими механизмами их разрушения).
В норме количество тромбоцитов 150-450/мкл, минимальный их уровень – критическая цифра Франка – 30/мкл (ниже нее можно ожидать спонтанных геморрагий).
Этиология идиопатической тромбоцитопенической пурпуры неизвестна, в основе патогенеза – фиксация на поверхности тромбоцитов IgG, направленных против АГ собственных тромбоцитов ® усиление фагоцитоза тромбоцитов макрофагами селезенки и печени ® повышенное разрушение тромбоцитов, укорочение продолжительности их жизни (с 7-10 дней в норме до нескольких часов)
Различают острую (обычно у детей 2-6 лет, длится не более 6 мес, характерны быстрое, внезапное начало, выраженный геморрагический синдром с последующим спонтанным выздоровлением или ремиссией) и хроническую (у врослых, длится несколько лет) формы заболевания.
Клиника хронической формы идиопатической тромбоцитопенической пурпуры.
- заболевание наиболее характерно для женщин, развивается постепенно, исподволь, носит хронический, рецидивирующий характер со сменой периодов обострения периодами ремиссии различной длительности
- геморрагический синдром:
а) кровоточивость петехиально-пятнистого типа в виде кожных геморрагий, локализующихся чаще всего на передней поверхности туловища, верхних и нижних конечностях, в местах инъекций; цвет геморрагических высыпаний меняется в зависимости от давности их появления: вначале пурпурно-красный, затем голубоватый, зеленоватый, желтый («цветение синяков»)
б) кровотечения из слизистых оболочек: носовые, десневые, полименорея, при тяжелом течении – почечные (макрогематурия), легочные (кровохарканье), желудочно-кишечные (мелена, рвота «кофейной гущей») и др. кровотечения
- могут наблюдаться внутримозговые и субарахноидальные кровоизлияния, кровоизлияния в склеру или сетчатку глаза, тяжелые кровотечения после тонзиллэктомии, экстракции зуба, во время операции и родов
- при частых и обильных кровотечениях - признаки постгеморрагической анемии (бледность кожи и видимых слизистых и др.)
Диагностика тромбоцитопенической пурпуры:
1. ОАК: снижение общего количества тромбоцитов < 100*109/л, их морфологические изменения (анизоцитоз, пойкилоцитоз и шизоцитоз); преобладают тромбоциты больших размеров (3-4 нм в диаметре),
встречаются тромбоциты малых размеров и фрагменты тромбоцитов («микрочастицы»); гипохромная анемия; умеренный нейтрофильный лейкоцитоз со сдвигом влево после обильной кровопотери
2. Исследование гемостаза: увеличение времени кровотечения (до 15 мин и более при норме 2,0-7,5 мин); нарушение ретракции кровяного сгустка
3. Иммунограмма: повышенное содержание IgG к АГ тромбоцитов, увеличение циркулирующих иммунных комплексов
Лечение тромбоцитопенической пурпуры:
1. Преднизолон или метилпреднизолон в начальной дозе 1 мг/кг/сут ® нет эффекта 5-7 дней ®
увеличение дозы до 2-3 мг/кг/сут (возможна пульс-терапия метилпреднизолоном); длительность гормональной терапии от 1-4 месяцев до полугода, геморрагии купируются в первые дни лечения, а тромбоциты увеличиваются постепенно
2. При неэффективности ГКС в течение полугода – спленэктомия
3. При неэффективности спленэктомии – химиотерапия (винкристин, азатиоприн, циклофосфамид в сочетании с преднизолоном)
4. Возможно применение больших доз человеческого Ig в/в (сандоглобулин 0,25 г/кг, затем поддерживающая доза 0,5 мг/кг каждые 15 дней) – иммуноглобулин закрывает рецепторы макрофагов и те перестают поглощать тромбоциты
5. Плазмаферез для удаления АТ
6. Курсы лечения дициноном (этамзилатом) по 1,5 г/сут внутрь в течение 14 дней
7. Инфузии тромбоцитов не показаны и используются только по жизненным показаниям
|
|
Дата добавления: 2014-01-04; Просмотров: 1427; Нарушение авторских прав?; Мы поможем в написании вашей работы!