КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Первичные иммунодефициты
|
|
|
|
ИММУНОДЕФИЦИТНЫЕ СОСТОЯНИЯ
Группу первичных иммунодефицитов образуют заболевания, в основе которых лежит наследственно обусловленная дефектность структуры и функционирования иммунной системы, которая проявляется в нарушении иммунной защиты.
Первичные иммунодефициты — это очень редкие состояния (примерно 1 больной на 1 000 000 человек). Они являются почти исключительно уделом детского возраста, поскольку значительная часть больных с тяжелыми формами иммунодефицитов не доживает до 20 лет, а при более легких формах иммунологические дефекты с возрастом в определенной степени компенсируются.
Как правило, в основе первичных иммунодефицитов лежит генетически обусловленный блок развития клеток иммунной системы или выпадение важных иммунных процессов вследствие дефекта определенных молекул, например ферментов или мембранных структур (схема 1).
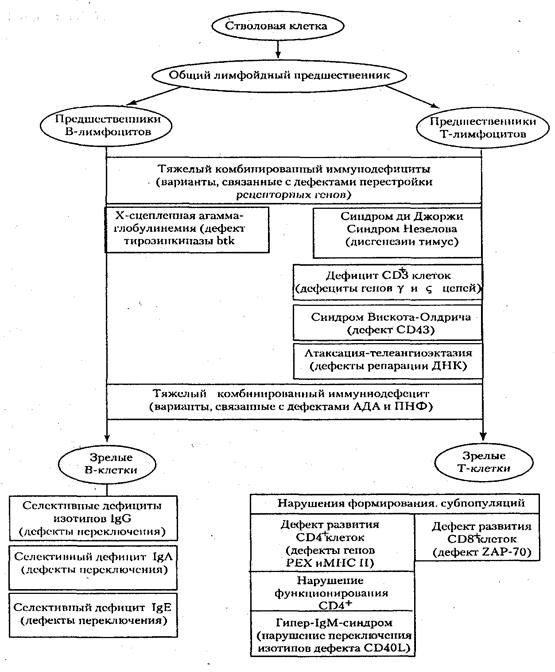
Первичные иммунодефициты можно разделить по преобладающему типу поражений звеньев иммунной системы на 3 типа:
— комбинированные иммунодефициты;
— иммунодефициты с преимущественным поражением клеточного иммунитета;
— преимущественно гуморальные иммунодефициты.
К первым относят заболевания, в основе которых лежат генетические дефекты, затрагивающие различные линии дифференцировки лимфоцитов, а также ранние этапы их развития, общие для Т- и В-линий. Во вторую группу входят иммунодефициты, при которых нарушается развитие Т-клеток и страдают опосредуемые ими реакции клеточного иммунитета; к этой же группе относятся дефекты фагоцитирующих клеток. В группу гуморальных иммунодефицитов включают патологию, в основе которой лежит нарушение развития В-клеток и Т-хелперов гуморального ответа, а также патологию компонентов комплемента.
В последние годы выясняются молекулярные основы поражения при первичных иммунодефицитах. Одной из первых была расшифрована природа комбинированных иммунодефицитов, связанных с недостаточностью ферментов пуринового метаболизма. Известны варианты таких дефектов, обусловленные мутациями генов, кодирующих аденозиндезаминазу и пуриннуклеотидфосфорилазу. Основой другой формы тяжелого комбинированного иммунодефицита, затрагивающего Т- и В-ростки лимфопоэза, служит дефект процесса перестройки генов антигенраспознающих рецепторов, связанный с отсутствием ферментов рекомбиназ, которые катализируют этот процесс.
Очень разнообразен спектр генетически обусловленных нарушений выработки антител. Их причиной может быть как поражение В-лимфоцитов (их развития или экспрессии генов иммуноглобулинов), так и дефектность Т-клеток (ослабление хелперной активности). Примером первого рода может служить агаммаглобулинемия Брутона, сцепленная сХ-хро-мосомой. Ее основой являются мутации гена, детерминирующего фермент тирозинкиназу btk, которая связана с антигенраспознающим рецептором В-лимфоцитов. Отсутствие этой тирозинкиназы делает невозможным развитие В-лимфоцитов уже на самых ранних стадиях.
В основе другого первичного иммунодефицита — гипер-IgМ-синдрома лежит дефект CD154 — молекулы, появляющейся на поверхности Т-клеток при их активации; в результате ее взаимодействия с молекулой CD40 поверхности В-лимфоцитов в эти клетки передается сигнал, обеспечивающий их дифференцировку в антителообразующие клетки, а также переключение изотипов секретируемых антител. В отсутствие этого сигнала происходит синтез иммуноглобулинов только одного изотипа — IgM, что сопровождается ослаблением гуморального иммунного ответа. Существуют формы гуморальных иммунодефицитов, при которых нарушено образование иммуноглобулинов какого-либо одного изотипа. Среди таких селективных дефектов наиболее частым является дефицит IgA.
При нем присутствуют В-лимфоциты, несущие мембранный IgA, однако не образуются плазматические клетки, секретирующие IgА-антитела.
Ряд комбинированных иммунодефицитов возникает при локализованных дефектах генов мембранных молекул адгезии. Следствием таких мутаций является нарушение миграции клеток, в первую очередь нейтрофилов и моноцитов/макрофагов, а также их взаимодействий с клетками других типов. Примером могут служить сходные поражения, развивающиеся как результат наследственных дефектов экспрессии (Р2-интегринов и углеводных детерминант, распознаваемых селектином L Эти поражения представляют собой два варианта LAD-синдрома — дефицит адгезии лейкоцитов, признаком которого является ослабление функции нейтрофилов, и повышение чувствительности к гнойным инфекциям.
Дефекты компонентов комплемента представлены вариантами с поражением практически всех основных факторов классического и альтернативного путей активации комплемента. Как правило, выпадение единичных компонентов системы комплемента проявляется в умеренном снижении устойчивости к некоторым возбудителям. Лишь дефицит ингибитора С1 q сопровождается развитием ангионевротического отека, обусловленного накоплением вазоактивных пептидов С5а и СЗа.
Иммунодефициты, в основе которых лежит дефект генов цитокинов, немногочисленны, что связано с «избыточностью» системы цитокинов, которая обусловлена взаимозаменяемостью их функций. Лишь когда генетический дефект затрагивает функцию многих цитокинов, это проявляется в тяжелых расстройствах иммунитета, что происходит, например, при дефекте гена уцепи, общей для рецепторов интерлейкинов 2, 4, 7, 13 и 15.
В результате дефекта, затрагивающего ген мембранного сиалопротеина CD43, развивается синдром Вискотта—Олдрича, о чем свидетельствует тромбоцитопения с геморрагическим синдррмом в сочетании с экземой и комбинированным иммунодефицитом. При этом заболевании аномально функционирует цитоскелет, что отражается на подвижности клеток и межклеточных взаимодействиях, важных для осуществления иммунных процессов.
При атаксии-телеангиэктазии наблюдается поражение различных функций, обусловленное слабостью аппарата репарации ДНК и нестабильностью хромосом, а также дефектами клеточного цикла. Это дает неожиданное сочетание симптомов: комбинированный иммунодефицит (недоразвитие вилочковой железы, дефицит Т-клеток и иммуноглобулинов «поздних» изотипов — IgG2, lgG4, IgE, IgA), неврологические отклонения (атаксия), поражение сосудистой стенки (телеангиэктазии), нарушение пигментации.
Помимо рассмотренных «точечных» поражений иммунной системы известны первичные иммунодефициты, развитие которых обусловлено множественными дефектами, затрагивающими формирование в эмбриогенезе различных органов, включая органы иммунной системы. Так, наследственный порок, приводящий к нарушению развития у эмбрионов человека производных 3 и 4 жаберных щелей, служит основой синдрома Ди Джорджи с дефектом развития вилочковой железы (она не заселяется предшественниками Т-клеток, развитие которых прерывается на костномозговой стадии) и гистогенетически родственных органов (паращитовидных желез и т.д).
Основным симптомокомплексом, отражающим нарушение иммунной защиты при первичных иммунодефицитах, является инфекционный синдром, т.е. понижение резистентности к инфекционным агентам, в том числе сапрофитным (Pneumocystis carinii, Candida, цитомегаловирус, некоторые энтеровирусы). Характер нарушений иммунной защиты определяется локализацией поражения в иммунной системе. Так, при блокаде процесса перестройки рецепторных генов отсутствуют как Т-, так и В-клетки и не развиваются ни клеточные, ни гуморальные формы иммунного ответа. При селективных дефектах определенных классов лимфоцитов, а также их субпопуляций выпадают именно те иммунологические функции, за которые ответственны поражаемые типы клеток. При блокаде развития В-клеток развивается агаммаглобулинемия с нарушением гуморальной защиты от внеклеточных бактерий и их токсинов, а при дефицитах Т-лимфоцитов страдает клеточная защита от вирусов и микобактерий. При некоторых формах первичных иммунодефицитов (атаксия-телеангиэкста-зия, синдром Вискотта-Олдрича и т.д.) значительно повышается риск развития злокачественных опухолей (до 10—15 %). Нередко нарушения иммунологических функций регистрируются при нормальной численности соответствующих клеток.
Клинико-иммунологическое обследование дает четкие результаты лишь при тех формах первичных иммунодефицитов, при которых точно локализован дефект. Так, при тяжелом комбинированном иммунодефиците отсутствуют как Т-, так и В-клетки, при синдроме Ди Джорджи резко снижено содержание Т-лимфоцитов, а при агаммаглобулинемиях — В-лимфоцитов. По изменению концентрации иммуноглобулинов в сыворотке или компонентов комплемента различных изотипов может быть установлена локализация дефекта в системе гуморального иммунитета. Все большую диагностическую значимость приобретает определение конкретных мембранных маркеров клеток иммунной системы (молекул адгезии, CD154, С043ит.д.), а также методы, позволяющие выявить мутации конкретных генов.
|
|
|
|
|
Дата добавления: 2013-12-13; Просмотров: 977; Нарушение авторских прав?; Мы поможем в написании вашей работы!