КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Кинетика кислотно-основного катализа
|
|
|
|
Скорость многих реакций, катализируемых кислотами и основаниями, определяется только кислотностью среды. В других случаях скорость может зависеть и от общей концентрации кислоты или основания, с помощью которых создается заданная кислотность. В водных растворах постоянное значение рН реакционной массы поддерживают обычно с помощью буферных растворов, представляющих собой раствор слабой кислоты АН или ее соли А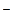 М+:
М+:
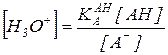
В зависимости от чувствительности скорости реакции к изменению общей концентрации буферного раствора при [Н3О+]= const или при [АН]/[А] = const, кислотно-основный катализ подразделяют на специфический и общий.
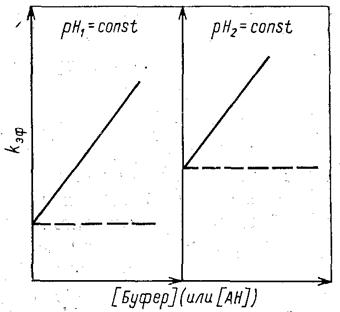
К специфическому относят такой тип катализа, при котором скорость реакции определяется только значением рН среды или концентрацией ионов Н3О+ или НОи не зависит от концентрации других кислот и оснований, присутствующих в реакционной массе. Типичный вид зависимостей скорости таких реакций от рН среды приведен на рис. 5.4. К общему катализу относят реакции, скорость которых зависит и от рН среды, и от концентрации и природы кислот (или оснований), находящихся в буферном растворе (рис. 5.5).
 | |||
 | |||
рН, Н0, Н- и т.п.
Рис. 5.4.Типичный вид зависимостей lgkэф Рис5.5. Кинетический тест на наличиеобщего
от функции кислотности среды или специфического кислотного катализа:
для реакций кислотно-основного катализа сплошные линии - общий кислотный катализ;
пунктир - специфический кислотный катализ
(рН1>рН2)
Специфический кислотный катализ может наблюдаться только при быстром установлении всех протолитических равновесий в растворе и последующем медленном превращении протонированного реагента в продукты реакции. Протолитические равновесия в реакционной массе при кислотном катализе включают реакции передачи протона между кислотой-катализатором АН, реагентом R и растворителем LH:
AH + R Û RH+ + A AH + LH Û LH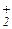 + A
+ A
LH+ R Û RH+ + LH
Одно из этих равновесий стехиометрически зависимо от других, и для количественной характеристики материального баланса системы можно ограничиться двумя последними. При этом степень протонирования реагента [RH+]/[R] однозначно определяется заданным значением h (или 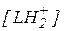 в разбавленных растворах):
в разбавленных растворах):
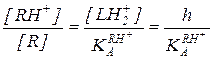 (5.46)
(5.46)
 Последующее превращение RH+ на лимитирующей стадии может протекать мономолекулярно - механизм А-1 [(5.47) или (5.48)], либо с участием второго исходного реагента Y - механизм А-2 (5.49).
Последующее превращение RH+ на лимитирующей стадии может протекать мономолекулярно - механизм А-1 [(5.47) или (5.48)], либо с участием второго исходного реагента Y - механизм А-2 (5.49).

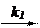 RH+ РН+ (5.47)
RH+ РН+ (5.47)
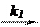 RH+R'H+ РН+ (5.48)
RH+R'H+ РН+ (5.48)
RH+ + Y PH+ (5.49)
Протонированный продукт реакции РН+ и второй исходный реагент Y, который всегда является нуклеофилом и обладает поэтому основными свойствами, также участвуют в протолитических равновесиях:

 P + LH2+ PH+ + LH Y + LH2+ YH+ + LH
P + LH2+ PH+ + LH Y + LH2+ YH+ + LH
По этим схемам осуществляется катализ реакций с участием реагентов, для которых характерны высокие скорости протолитических реакций, т. е. реагентов, протонирующихся по атомам О, N, S и F. Согласно приведенным схемам, скорость реакции описывается кинетическими уравнениями:
A-1: v = k1 [RH+] (5.50)
A-2: v = k1 [RH+][Y] (5.51)
Они включают концентрации протонированной формы реагента [RH+] и свободной формы нуклеофильного реагента [Y]. Для практического использования необходимо преобразовать эти уравнения в форму, в которой [RH+] и [Y] выражены через аналитические (суммарные) концентрации CR и CY и через известную концентрацию [+LН2] или h. Уравнения (5.50) и (5.51) имеют наиболее простой вид при постоянстве h[+SН2] в ходе реакции. Это условие выполняется:
- при близкой и малой основности продукта Р и исходных реагентов Y и R, так что превращение
Y + R ® P
не изменяет кислотности среды;
- при проведении реакции в буферном растворе или в концентрированном растворе кислоты АН, взятой в большом избытке по отношению к R и Y.
Преобразование кинетического уравнения (5.50) для таких условий приводит его к виду:
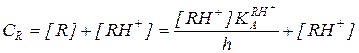
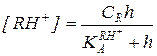
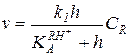 (5.52)
(5.52)
При постоянстве h[+LН2] зависимость CR от времени описывается кинетическим уравнением первого порядка
v=kэфCR,
в котором константа скорости есть функция кислотности реакционной массы:
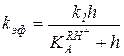 (5.53)
(5.53)
В логарифмических координатах (см. рис. 5.4) это уравнение соответствует кривой ж.
Действительно, при малой кислотности и слабых основных свойствах реагента (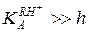 ) уравнение (5.53) принимает вид:
) уравнение (5.53) принимает вид:
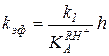 или
или 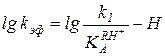 , (5.54)
, (5.54)
что соответствует правому линейному участку кривой ж или зависимости, представленной на рис. 5.4, а.
При высокой кислотности среды и достаточной основности реагента (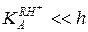 ) в знаменателе уравнения (5.53) можно пренебречь
) в знаменателе уравнения (5.53) можно пренебречь 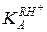 , и экспериментально определяемая константа скорости не будет зависеть от кислотности среды k эф = k1. Это означает протонирование реагента, когда дальнейшее возрастание кислотности уже не увеличивает концентрацию RH+ и kэф (горизонтальный участок кривой рис. 5.4, ж). Криволинейный участок этой зависимости отвечает соизмеримым значениям и h и описывается уравнением (5.53), которое приводится к линейному виду относительно неизвестных параметров k 1 и :
, и экспериментально определяемая константа скорости не будет зависеть от кислотности среды k эф = k1. Это означает протонирование реагента, когда дальнейшее возрастание кислотности уже не увеличивает концентрацию RH+ и kэф (горизонтальный участок кривой рис. 5.4, ж). Криволинейный участок этой зависимости отвечает соизмеримым значениям и h и описывается уравнением (5.53), которое приводится к линейному виду относительно неизвестных параметров k 1 и :
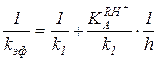 (5.55)
(5.55)
Это позволяет по экспериментальной зависимости 1/kэф от 1/h определить константу скорости лимитирующей стадии k1 и константу кислотности .
Уравнением (5.54) описываются реакции третичных спиртов и их эфиров, реакции гидролиза и алкоголиза уксусного ангидрида и т. п. Эти реакции ускоряются небольшими добавками сильных кислот; скорость реакций пропорциональна кислотности (h0, [Н3О+]) и не зависит ни от концентрации, ни от природы нуклеофильного реагента. Скорость реакций, протекающих по механизму А-1 в концентрированных растворах кислот, описывается уравнением (5.52) в его общей форме, ему соответствует кривая рис. 5.4, ж.
Преобразование кинетического уравнения реакции, протекающей по механизму А-2 (5.51), путем выражения концентраций RH+ и свободного реагента Y через суммарные концентрации этих реагентов дает:
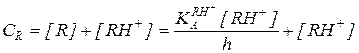
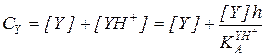
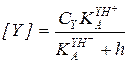
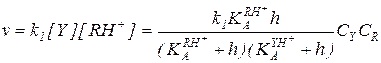 (5.56)
(5.56)
Из уравнения (5.56) следует, что при h = const зависимость концентраций реагентов от времени для реакции, протекающей по механизму А-2, описывается кинетическим уравнением второго порядка, в котором экспериментально определяемая константа скорости зависит от кислотности так:
 (5.57)
(5.57)
Зависимость (5.57) в логарифмических координатах (см. рис. 5.4) дает кривую е с максимумом при
 .
.
При кислотности, существенно меньшей hмакс, когда h << 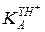 и h << , уравнение (5.57) принимает вид:
и h << , уравнение (5.57) принимает вид:
или
Ему соответствует правая нисходящая ветвь кривой рис. 5.4, е. В этих условиях степень протонирования как R, так и Y незначительна, суммарная концентрация CY практически равна концентрации свободного реагента Y, a [RH+] возрастает прямо пропорционально h.
При высоких значениях кислотности (h >>и h >> ) уравнение (5.57) преобразуется в форму:
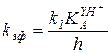 или
или 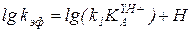 , (5.58)
, (5.58)
которая описывает снижение скорости на левой нисходящей ветви кривой е. При столь высокой кислотности реагенты R и Y практически полностью протонированы и снижение скорости связано с уменьшением незначительной доли еще не протонированного свободного реагента Y, концентрация которого обратно пропорциональна h.
Наличие максимума скорости характерно для многих реакций, протекающих по механизму А-2 в концентрированных растворах кислот. К ним относятся, в частности, реакции нитрования ароматических соединений, механизм которых можно представить схемой
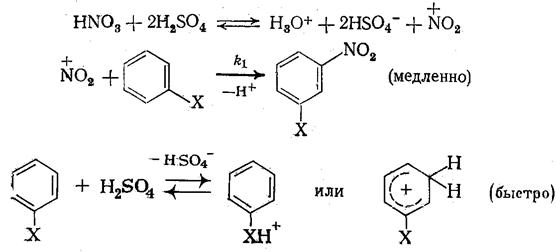
Возрастание скорости с увеличением кислотности при низких и умеренных концентрациях кислоты-катализатора (H2SO4) обусловлено повышением равновесной концентрации +NO2, а снижение скорости в области высоких кислотностей - протонированием второго реагента (АrН) и превращением его в неактивную форму при практически полном превращении HNO3 в +NO2.
Такие же экстремальные зависимости характерны для гидролиза некоторых амидов и сложных эфиров.
При гидролизе менее основных реагентов, характеризующихся большим значением , максимум скорости смещен в сторону очень высокой кислотности, а иногда совсем не проявляется. В частности, скорость гидролиза нитрилов возрастает во всем диапазоне увеличения кислотности. Этот факт используют для регулирования селективности гидролиза нитрилов. Если требуется остановить гидролиз на стадии образования амида, проводят реакцию при высоких концентрациях кислоты, правее максимума скорости гидролиза амида. В таких условиях при высокой скорости гидролиза нитрила образующийся амид практически не гидролизуется. И, наоборот, для гидролиза нитрилов до соответствующих кислот предпочтительнее проводить реакцию при низких концентрациях кислоты, когда амид гидролизуется быстрее, чем нитрил.
Кинетическое описание реакций кислотно-каталитического гидролиза, протекающих по механизму А-2, имеет некоторые особенности из-за неопределенности кинетического порядка по воде и отсутствия аналитического выражения для концентраций свободной воды. По этой причине для обработки экспериментальных зависимостей скорости гидролиза от кислотности среды используют известные данные по активности воды (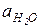 ) в растворах кислот. При низкой основности реагента или при умеренной кислотности, когда [RH+] = (h / )Cr и скорость реакции возрастает с кислотностью, кинетическое уравнение реакции А-2 можно представить в виде:
) в растворах кислот. При низкой основности реагента или при умеренной кислотности, когда [RH+] = (h / )Cr и скорость реакции возрастает с кислотностью, кинетическое уравнение реакции А-2 можно представить в виде:
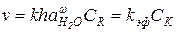 (5.59)
(5.59)
После логарифмирования выражения для константы скорости получим:
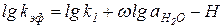 (5.60)
(5.60)
Кинетический порядок по воде ω в уравнениях (5.59) и (5.60) характеризует число молекул воды, необходимое для превращения протонированного реагента RH+ в активированный комплекс, причем одна из этих молекул играет роль нуклеофила, а другие специфически сольватируют активированный комплекс.
Рассмотренные виды зависимостей константы скорости от кислотности не исчерпывают возможных вариантов зависимостей в координатах lgkэф - Н (рН). В частности, для гидролиза некоторых сложных эфиров (например, этилацетата) получена зависимость, представленная кривой и (см. рис. 5.4). Правая ее часть с максимумом скорости характерна для рассмотренных реакций, протекающих по механизму А-2. Однако дальнейшее увеличение кислотности и переход в область высококонцентрированных растворов серной кислоты вновь приводит к повышению скорости реакции, которое связано с изменением механизма от А-2 к А-1.
Еще более разнообразны зависимости скорости от кислотности для многостадийных реакций, подверженных разному типу катализа на различных стадиях. Изменение кислотности среды в этих случаях может по-разному влиять на скорость последовательных стадий, изменять лимитирующую стадию и приводить к изломам и появлению экстремумов на кривых в координатах lgkэф - Н (см., например, кривые з, г, д на рис. 5.4).
Специфический основный катализ распространен значительно меньше, чем кислотный. Его схема включает быстрые протолитические реакции реагента RH с основанием-катализатором Ви лиат-ионом L.
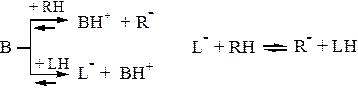
и последующее медленное превращение активированного реагента R-, которое происходит мономолекулярно или при участии второго реагента:
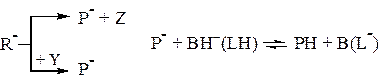
Скорость реакций, протекающих по этой схеме, однозначно определяется концентрацией лиат-иона (L) или рН (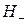 ) реакционной массы, и кинетические закономерности реакций имеют тот же вид, что и для специфического кислотного катализа.
) реакционной массы, и кинетические закономерности реакций имеют тот же вид, что и для специфического кислотного катализа.
При общем кислотно-основном катализе скорость реакции зависит от концентрации каждой формы кислотного или основного катализатора. Такие кинетические закономерности наблюдаются, если стадия отрыва протона от реагента основанием-катализатором или стадия присоединения протона к реагенту от кислоты-катализатора является лимитирующей. Механизм реакций можно представить схемой:
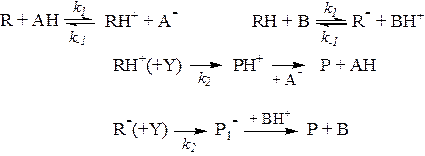
Образовавшийся на первой стадии активированный реагент (Rили RH+) далее быстро превращается в продукты по мономолекулярной реакции или при участии второго реагента Y. При условии, что k 2 [Y] >> k-1[A-(BH+)] и k2[Y]>> k1[R(RH)], весь процесс лимитируется скоростью протолитической реакции, икинетическое уравнение имеет вид:
v = К [R] [АН] или v = k1 [RH] [В]
При этом для взаимодействия каждой формы катализатора среагентом есть своя константа скорости, и в общем виде последнее уравнение можно представить в форме:
 или
или 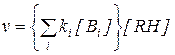 (5.61)
(5.61)
Рассматриваемые схемы характерны для реакций, в которых на первой стадии происходит медленный разрыв или образование связей С–Н. В качестве примера можно привести кислотно-каталитические реакции олефинов, протекающие через-промежуточное образование ионов карбония, и основно-каталитические реакции енолизации кетонов:
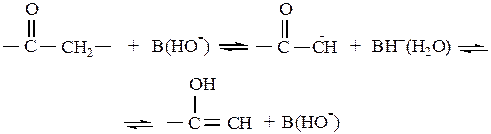
Общий кислотно-основный катализ проявляется, однако, не только при разрыве или образовании связей С–Н на стадии активирования реагента. Известно множество примеров, когда лимитирующей оказывается стадия передачи протона между атомами О–О, О–N, N–N и др. Это происходит в случаях, когда перенос протона сопровождается синхронным разрывом или образованием новой связи. Синхронному процессу обычно предшествует предварительная координация кислоты и основания за счет образования водородной связи. Общая схема такого процесса имеет вид:
a) HA + R 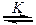 R×××HA B + RH B×××HR
R×××HA B + RH B×××HR
б) R×××HA  Продукты B×××HR Продукты
Продукты B×××HR Продукты
в) R×××HA + Y Продукты B×××HR + Y Продукты
Равновесие образования комплекса с водородной связью между атомами О, N, S, F и С1 всегда устанавливается быстро. Последующий перенос протона сопровождается одновременным изменением других связей в R или RH и лимитирует скорость процесса. Эта лимитирующая стадия может быть мономолекулярной (б) или бимолекулярной (в). Регенерация исходной формы катализатора происходит на этой же стадии или в результате быстрых последующих превращений. Примерами бимолекулярных превращений (в) активированного реагента при кислотном катализе являются этерификация метанола уксусной кислотой и метанолиз трифенилхлорметана, а при основном катализе – реакции аминов с производными карбоновых кислот. Примером мономолекулярного превращения (б) комплекса катализатора с реагентом является гидролиз орто -эфиров.
Схема общего кислотного катализа с бимолекулярной лимитирующей стадией аналогична рассмотренной выше схеме нуклеофильного катализа в реакциях присоединения. Они различаются лишь тем, что при нуклеофильном катализе второй реагент Y представляет собой нуклеофильный катализатор, а кислота-катализатор НА является реагентом. Кинетический анализ последовательности элементарных стадий (а) и (в) приводит поэтому к тем же кинетическим уравнениям. Кинетическое уравнение лимитирующей стадии:
v = k 1[ R∙∙∙ HA][Y] (5.62)
С учетом комплексообразования, при избытке реагента R по отношению к катализатору уравнение (5.62) принимает вид:
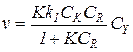 (5.63)
(5.63)
Важным вопросом в общем кислотно-основном катализе является установление связи каталитической активности кислоты или основания с их строением. Если при специфическом катализе скорость реакции однозначно определяется кислотностью реакционной массы (рН, H  ), то при общем кислотно-основном катализе значения экспериментально определяемых констант скорости (k1, K) являются функцией природы кислоты или основания-катализатора.
), то при общем кислотно-основном катализе значения экспериментально определяемых констант скорости (k1, K) являются функцией природы кислоты или основания-катализатора.
Кислотно-основный катализ нередко сопровождается некаталитической реакцией. Иногда реакция катализируется как кислотой, так и основанием. Существуют процессы, в которых кислотность среды изменяется или за счет больших различий в основности реагентов и продуктов, или из-за расходования кислоты, когда она одновременно является и реагентом, и катализатором (сульфирование ароматических соединений, сульфатирование спиртов).
|
|
|
|
Дата добавления: 2014-01-07; Просмотров: 4733; Нарушение авторских прав?; Мы поможем в написании вашей работы!