КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Механизм реакций
|
|
|
|
Термодинамика и кинетика реакций гидроочистки.
Сернистые соединения нефтей являются сложными смесями, состоящими из меркаптанов, сульфидов (с открытой цепью и циклических), а также дисульфидов и гетероциклических соединений. Групповой состав сернистых соединений нефтей весьма сильно различается. Помимо элементарной серы и сероводорода в сырых нефтях идентифицировано 111 сернистых соединений [1].
Связь С – S менее прочна, чем связь С – С. Однако прочность связи нужно оценивать с учетом компенсации энергии, идущей на ее разрыв, и энергии образования новой связи с катализатором в переходном комплексе. Например, на никеле энергия разрыва связей С – С, С – N и C – S составляют (в ккал/моль):
С – С 48,8
С – N 26,0
C – S 5,0
Термодинамика реакций деструкции сернистых соединений очень благоприятна: все реакции тиолов, алифатических и циклических сульфидов имеют положительные значения логарифма константы равновесия до 900 К. Только константа равновесия реакции
 ® С4Н10 + Н2S
® С4Н10 + Н2S
быстро уменьшается с ростом температуры, ее логарифм достигает нуля при 5400С, но в температурных пределах гидроочистки и она имеет положительные значения логарифма константы равновесия (рис. 1.1) [1].
В условиях гидроочистки сернистые соединения алифатического ряда настолько нестабильны, что термодинамически вероятен их распад на сероводород и олефин, а также образование дисульфидов:
2RSH 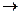 R – S – R + H2S
R – S – R + H2S
Поскольку логарифм константы равновесия реакции циклизации бутантиола
C4H9SH + 3H2
при 427 0С равен примерно +0,6, реакции циклизации могут протекать при термическом воздействии на нефть. Этим объясняется тот факт, что нефтепродукты вторичного происхождения обычно более богаты тиофенами, чем продукты первичного происхождения.
Механизм реакций гидрообессеривания, или гидродесульфирования (ГДС) чаще всего изучают на примере превращений тиофена. Это объясняется тем, что тиофен и его производные – наименее активные сернистые соединения нефти. Предложены схемы реакций тиофена и его производных на алюмокобальтмолибденовых катализаторах в условиях гидроочистки (рис. 1.2).
Предположение о том, что первой реакцией тиофена по первому реакционному пути является расщепление связи C-S, а не гидрирование связи С=С, подтверждается следующим экспериментальным фактом:
| гидрированное соединение с незатронутыми связями C-S (тетрагидротиофен) дает продукты гидрообессеривания, отличные от тех, которые наблюдаются для тиофена.
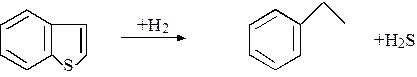
Изучались также реакции, протекающие при обессеривании бензотиофена. Наблюдалось установление равновесия в реакции гидрирования – дегидрирования с более высокой скоростью по сравнению со скоростью обессеривания:
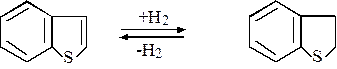 Скорости обессеривания обоих соединений были примерно одинаковы, поэтому не удалось установить, является ли одно промежуточным продуктом при обессеривании другого.
Скорости обессеривания обоих соединений были примерно одинаковы, поэтому не удалось установить, является ли одно промежуточным продуктом при обессеривании другого.
|
| Р и с. 1.1. Зависимость константы равновесия реак-ции восстановления сернис-тых соединений водородом с образованием насыщен-ных углеводородов и серо-водорода от температуры: 1- этантиол; 2 – тиацик-логексан; 3 – 2-тиабутан; 4 – тиофен; 5 – 3,4-дитиагексан |
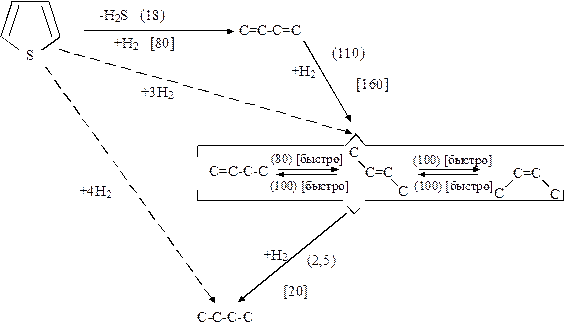 Р и с. 1.2. Схема реакции гидрообессеривания тиофена:
Р и с. 1.2. Схема реакции гидрообессеривания тиофена:
числа в скобках – приблизительные скорости [ммоль/(г·с)]; в круглых скобках для катализатора Cr2O3 при 415оС, в квадратных скобках – для катализатора СоМо/А12О3 при 400оС
Механизм реакции бензтиофена:
Гидрообессеривание дибензтиофена происходит с высокой селективностью как при низких, так и при высоких давлениях:
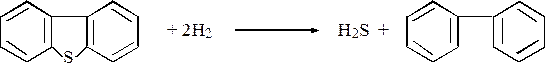
Подробное представление реакционной схемы, включающей дибензотиофен и водород в присутствии сульфидированного СоО – МоО3/γ-А12О3 при 300оС и 10 МПа, дано на рис. 1.3.
Конверсия дибензтиофенов зависит от положения и числа заместителей. Наличие заместителей в положениях, ближайших к S, значительно уменьшает реакционную способность, т.к. они создают стерические препятствия адсорбции атома серы на активной поверхности:
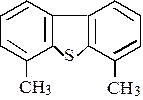
Метильные заместители в других положениях мало влияют на реакционную способность молекулы дибензтиофена(рис. 1.4 и 1.5).
На алюмоникельмолибденовых (АНМ) катализаторах выше вероятность гидрирования ароматических колец, являющихся частью молекулы дибензтиофена, чем на алюмокобальтмолибденовых (АКМ) катализаторах. Гидрирование вызывает деформацию жестко плоскостной структуры дибензтиофена и облегчает адсорбцию атома серы на поверхности катализатора.
Тщательное количественное изучение кинетики обессеривания, охватывающее широкий диапазон температур и давлений и включающее влияние всех реагентов и продуктов, не проведено
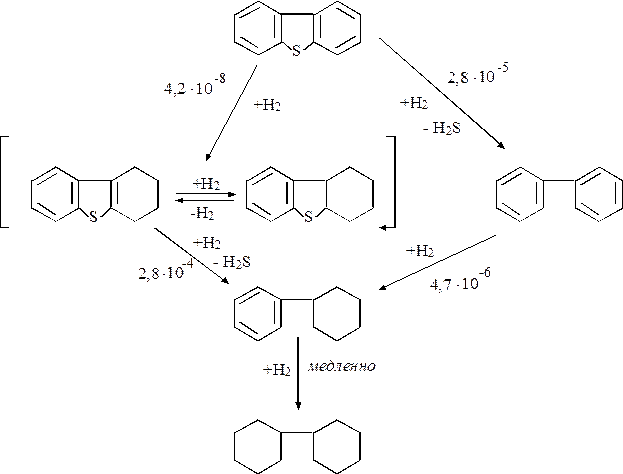
Р и с. 1.3. Схема реакции гидрообессеривания дибензтиофена: катализатор – сульфидированный СоМо/А12О3 при 300оС и давлении 10,2 МПа; числа над стрелками – константы скорости реакции псевдопервого порядка (в м3/кг катализатора·с)
исчерпывающим образом [2, с.491]. Частичные кинетические исследования гидрообессеривания тиофена при атмосферном давлении и температуре от 235 до 2650С на промышленном катализаторе гидроочистки в дифференциальном реакторе, проведенные Сеттерфилдом, показали, что реакция ингибируется сероводородом [3, с.337]. Данные о скоростях исчезновения тиофена (гидрогенолиза) и образования бутана (гидрирования бутена) были представлены уравнениями скорости Лэнгмюра – Хиншельвуда
rгидрообес = 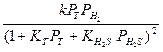
rгидрир =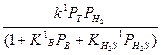
Индекс Т в этих уравнениях означает «тиофен», В – «бутен».
Применимость этих уравнений точно не установлена с помощью полученных данных. Несколько важных качественных выводов подтверждается опубликованными данными: 1) H2S ингибирует как гидрогенолиз, так и гидрирование; 2) значительные количества тиофена и бутена адсорбируются на поверхности катализатора конкурентно с H2S; 3) менее ясен, но более важен вывод о том, что реакции гидрогенолиза и гидрирования происходят на разных каталитических центрах.
Очень важной реакцией, протекающей в условиях гидроочистки, является гидрирование ароматических углеводородов. На обычных промышленных катализаторах гидроочистки в основном протекают реакции частичного гидрирования конденсированных ароматических углеводородов.
Механизм гидрирования ароматических углеводородов на сульфидированных АНМ катализаторах включает в себя реакцию образования промежуточного p - комплекса. Он образуется ионом переходного металла на поверхности сульфидного катализатора и p - электронами ароматического кольца. Ион переходного металла представляет собой льюисовский кислотный центр (L – центр), являющийся акцептором электронов.
Ароматическая связь трудно гидрируется вследствие резонансной стабилизации двойных связей в ароматическом кольце. Образование при адсорбции ароматического соединения p - комплекса между активным центром катализатора и электронами ароматического кольца ослабляет связи в кольце и делает их более уязвимыми для атаки водорода. Рост электроноакцепторной способности, или кислотности активной фазы катализатора гидроочистки способствует гидрированию.
Обе реакции – гидрогенолиз связи C-S и гидрирование ароматических углеводородов – отравляются органическими основаниями, но в разной степени. Снижение скорости этих реакций в результате отравления происходит, соответственно, в 5 и в 1000 раз. Следовательно, активные центры этих реакций похожи и различаются только кислотностью.
Многочисленными исследованиями показано, что кислотность АНМ и алюмокобальтмолибденовых (АКМ) катализаторов связана с сульфидной фазой. Кислотные центры являются льюисовскими. Это показано ИК - спектроскопическими исследованиями. Высказано обоснованное предположение о связи кислотных центров с анионными вакансиями на поверхности активного сульфида молибдена.
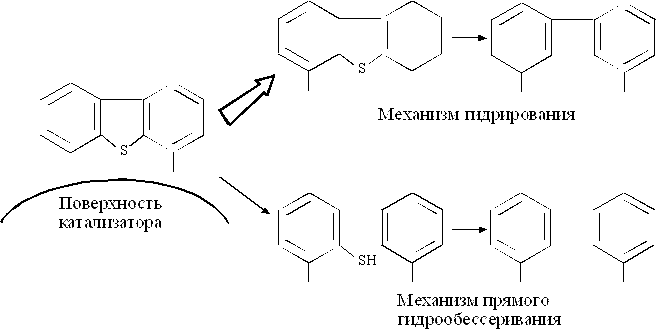
Р и с. 1.4. Преобладающий механизм реакции превращения
4,6 – диметилдибензтиофена

| Прямое гидрообессеривание >гидрирование Прямое гидрообессеривание >гидрирование Прямое гидрообессеривание <гидрирование Прямое гидрообессеривание <<гидрирование |
| Р и с. 1.5. Преобладающий механизм реакции в зависимости от положения алкильной группы |
|
|
|
|
|
Дата добавления: 2014-01-14; Просмотров: 2607; Нарушение авторских прав?; Мы поможем в написании вашей работы!