КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Качественный анализ. Обработка хроматограмм
|
|
|
|
ПРАКТИЧЕСКАЯ ЧАСТЬ
Обработка хроматограмм.
При выходе из хроматографической колонки компоненты разделяемой смеси поступают в детектор и фиксируются в виде хроматограммы. Последняя может служить основой качественного и количественного анализа.
Существует несколько способов качественного анализа разделенной в хроматографической колонке смеси: по характеристикам удерживания; с использованием аналитических методов или графических зависимостей между характеристиками удерживания веществ и их строением, свойствами и условиями анализа; а также с использованием индексов удерживания.
Время удерживания и пропорциональный ему удерживаемый объем могут быть положены в основу качественной идентификации веществ. Для качественной характеристики могут применяться как абсолютные, так и относительные величины удерживания. Поскольку на эти характеристики оказывает влияние объемная скорость газа-носителя, температура колонки, длина и диаметр колонки, толщина пленки жидкой фазы и ее природа, то идентификацию компонентов можно проводить только при постоянстве указанных условий.
Другой вариант анализа заключается в том, что в анализируемую смесь добавляется соединение (метод добавки), наличие которого предполагается.
Увеличение высоты пика одного из компонентов (без заметного увеличения его ширины) может свидетельствовать о наличии в смеси искомого соединения. Появление нового пика указывает на отсутствие данного соединения в смеси. Однако для окончательной идентификации рекомендуется провести подобный анализ на нескольких фазах различной полярности. Метод прост, но необходимо большое количество эталонных соединений при установлении состава смеси, состоящей из большого числа соединений.
Графические методы идентификации. В практике качественного хроматографического анализа веществ, относящихся к одному гомологическому ряду, широко применяются линейные зависимости логарифмов приведенных (относительных) времен (объемов) удерживания гомологов (lg t’; lg V’) от числа атомов углерода:
lg t’ = a1 + b1 nZ,
где a1 и b1 - константы, а nZ - число атомов углерода в молекуле (или молекулярная масса).
Аналогичная зависимость может быть получена и для температуры кипения гомологов:
lg t’ = a2 + b2 Tкип.,
или отношения температуры кипения компонентов к температуре колонки:
lg t’ = a3 + b3 Tкип / Tкол,
где a2, b2 и a3, b3 - константы, Ткип - температура кипения неизвестного соединения (или гомологов), Ткол - температура колонки (термостата).
Если для нескольких представителей одного гомологического ряда построить график, то получается прямая линия. Для другой неподвижной фазы или другого гомологического ряда получается прямая с другим наклоном. По известной величине времени (или объема) удерживания неизвестного соединения можно легко определить число атомов углерода в его молекуле или его температуру кипения и тем самым отождествить его с каким-либо известным соединением.
Однако в ряде случаев наблюдается отклонение от линейной зависимости, особенно для гомологического ряда сильно полярных веществ.
Значительно более удобными для идентификации веществ оказались индексы удерживания, введенные Ковачем. Этот способ получил в последнее время наибольшее применение. Суть этого метода заключается в использовании линейной зависимости между логарифмами объемов удерживания и числом атомов углерода нормальных парафинов как шкалы индексов I (индекс удерживания Ковача).
В этой шкале индексы удерживания нормальных углеводородов равны числу углеродных атомов, умноженному на 100. Идентификация с помощью индексов Ковача производится путем сравнения полученных значений Ia с табличными. Для этого получают хроматограмму анализируемого вещества и двух парафиновых углеводородов, один из которых выходит до, а другой - после анализируемого компонента. Индексы удерживания, как и относительные объемы (времена) удерживания, не зависят от неточностей в измерении расхода подвижной фазы, массы сорбента в колонке. Они сравнительно мало чувствительны к изменению температуры. При их использовании не требуется введения поправки на перепад давления (давление газа-носителя на входе и выходе из колонки).
Изотермический индекс удерживания соединения рассчитывается по формуле, в которую входят приведенные времена (объемы) удерживания двух соседних с ним членов гомологического ряда:
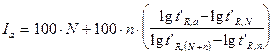 (15),
(15),
где N - число атомов углерода в н-алкане с более короткой цепочкой; n - разность в числе атомов углерода между двумя алканами, используемыми как стандарты.
Индексы Ковача зависят лишь от физико-химических свойств вещества, природы неподвижной жидкой фазы и температуры колонки и могут быть поставлены в ряд с такими константами химических соединений, как температура кипения, показатель преломления, плотность.
Количественный анализ.
Хроматографические методы широко применяются для количественного анализа газообразных и жидких проб. Измерение концентрации при помощи современных хроматографов основано на пропорциональности между количеством вещества и высотой или площадью хроматографического пика (в линейном диапазоне работы детектора).
Количественный анализ состоит из следующих этапов:
· отбор и подготовка проб к анализу;
· введение пробы в хроматографическую систему;
· хроматографирование и снятие хроматограммы;
· обработка результатов хроматографического анализа.
В хроматографическом анализе чаще применяются дифференциальные детекторы, поэтому рассмотрим методы расчета по дифференциальным хроматограммам.
Они состоят из ряда последовательно расположенных пиков, число которых должно соответствовать числу компонентов анализируемой смеси. Это условие может не выполняться, если в хроматографической колонке не происходит разделения двух или нескольких соседних компонентов. Взаимное наложение пиков нескольких компонентов может привести к ошибочному расчету количественного состава смеси. Поэтому очень важно предварительно проводить идентификацию всех компонентов смеси и знать ее полный качественный состав.
Вторым условием количественного анализа является полное поступление в колонку всех компонентов введенной для анализа пробы, и ее адекватное (полное) испарение.
Наконец, экспериментатор должен быть уверен, что ни одно из анализируемых веществ не вступает в необратимую реакцию с неподвижной фазой, сорбентом или твердым носителем, материалом, из которого изготовлены колонка, а также в том, что все вещества регистрируются детектором.
Задачей количественного газо-хроматографического анализа может быть:
1. определение индивидуального состава многокомпонентной смеси (при этом число пиков на хроматограмме должно соответствовать числу компонентов смеси, т.е. должно быть полное разделение смеси), (примером может служить определение индивидуального состава
2. бензиновой фракции, при определении октанового числа хроматографическим методом);
3. определение содержания одного или нескольких компонентов смеси (в этом случае достаточно, чтобы произошло разделение определяемых компонентов, а пики остальных могут накладываться), (например, определение фенола и монозамещенных алкилфенолов в сточной воде при производстве фенол содержащих полимеров).
Для количественной характеристики могут служить как высота пика hi, так и его площадь Si. Иногда для этих целей используют произведение высоты на время удерживания (h t). Выбор того или иного параметра для расчета зависит, главным образом, от формы хроматографических пиков и их взаимного расположения.
Измерение площади или высоты хроматографических пиков может привести к существенным погрешностям и оказать влияние на точность количественного анализа. Поэтому ко всем методам измерения площади (высоты) предъявляются следующие требования: они должны обеспечить хорошую воспроизводимость измерений даже в случае частично перекрытых пиков; каждое измерение должно выполняться не
менее 3-5 раз с тем, чтобы иметь возможность пользоваться средними величинами; измеряемые величины должны линейно зависеть от концентрации или потока вещества во всем диапазоне измерений.
Наиболее простым является измерение высоты пика. Однако, для пиков размытых и не имеющих острой вершины измерение высоты может привести к значительным погрешностям. Следует отметить, что диапазон линейной связи высоты пика с величиной вводимой пробы меньше (по сравнению с такой связью для площадей пиков). Поэтому измерения по высотам пиков производят в тех случаях, когда количество вводимой пробы не превышает 100 мкг для насадочных и 0,1 мкг - для капиллярных колонок.
Площади пиков в меньшей степени зависят от условий эксперимента, кроме того, измерение площадей дает большую точность. Эти обстоятельства явились причиной преимущественного распространения количественных измерений по площадям пиков.
Для измерения площади пика используют следующие способы:
1. графический метод, основанный на геометрических приемах измерения площади треугольника или трапеции на анализе и преобразовании уравнения гауссовской кривой;
2. автоматическое измерение интеграторами или ЭВМ с использованием специальных программ для количественной обработки хроматограмм.
Расчет площади пика по методу треугольника. Пик рассматривают как треугольник и его площадь рассчитывают как площадь треугольника.
а) 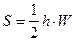 б)
б) 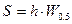 c)
c) 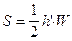 ,
,
где S - площадь пика; h - его высота; h’ - высота, измеренная от нулевой линии до точки пересечения касательных к сторонам пика; W - ширина пика у его основания, измеренная между точками пересечения с касательными к его сторонам; W0.5 - ширина пика на половине его высоты.
Рассчитанная этими способами площадь пика составляет около 80%, 94% и 97%, соответственно. На практике чаще всего используют формулу для расчета площади пика измеряя ширину пика на половине его высоты (б).
Методы количественного расчета. Чтобы провести количественный расчет хроматограммы, зная площади пиков, можно воспользоваться одним из следующих методов:
· абсолютной калибровки;
· нормализацией или нормализацией с калибровочными коэффициентами;
· внутренней стандартизацией.
1. Количественный анализ по методу абсолютной калибровки. Этот метод является наиболее точным и поэтому наиболее пригодным при определении микропримесей, а также в тех случаях, когда нужно определить лишь отдельные компоненты смеси. Метод состоит в установлении строгой числовой зависимости между сигналом детектора (площадь или высота пика) и количеством интересующего вещества, т.е. заключается в построении графика “площадь пика - количество вещества в пробе”. Если зависимость линейная, то определяют угловой коэффициент прямой Ki (абсолютный калибровочный коэффициент):
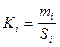
где Ki - абсолютный калибровочный коэффициент для компонента i; mi - масса компонента; Si - площадь пика, соответствующая этому компоненту.
Зная калибровочный коэффициент содержание вещества в пробе определяется по формуле:
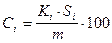
где Ci - содержание компонента i в пробе, масс. или об. %; Si - площадь его пика; m - масса пробы; V - объем пробы; Ki - абсолютный калибровочный коэффициент (коэффициент пропорциональности).
При выполнении анализа методом абсолютной калибровки необходимо строго соблюдать постоянство условий анализа и калибровки, а также должна быть реализована высокая точность и воспроизводим ость в дозировании пробы.
Недостаток метода - приходится строить калибровочные графики для каждого индивидуального вещества. Метод очень трудоемкий и требует наличия чистых реагентов для калибровки.
Количественный анализ методом нормализации. Метод основан на предположении, что вещества, независимо от их строения, взятые в одинаковом количестве, дают одну и ту же площадь пика.
Т.е. детектор будет одинаково чувствителен ко всем компонентам смеси, что бывает при анализе очень близких по свойствам веществ (одного класса). Для количественного анализа вначале суммируют площади всех пиков и делят площадь каждого отдельного компонента на сумму площадей. Процентное содержание компонента в анализируемой смеси рассчитывают по формуле:
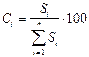
где Ci - содержание компонента i в пробе, масс.% или об.%; Si - площадь его пика.
Но этот метод не дает точных результатов в случае различной чувствительности выбранного детектора по отношению к разделяемым компонентам. В этом случае пользуются методом нормировки с калибровочными коэффициентами fi, т.е. учитывают различие в чувствительности детектора к различным компонентам смеси:
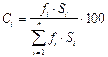
где Ci - содержание компонента i в пробе, масс. или об. %; Si - площадь его пика; fi - калибровочный коэффициент.
Наиболее распространенные способы определения калибровочных коэффициентов - метод абсолютной калибровки или относительно какого-нибудь стандарта.
Относительные поправочные коэффициенты рассчитывается по формуле:
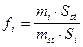
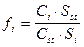
где fi - относительный поправочный коэффициент для компонента i; fst - поправочный коэффициент стандарта равен 1; Si - площадь пика компонента i; Sst - площадь компонента, принятого за стандарт; mi и Ci - масса или концентрация компонента i (масс.% или об.%); mst и Cst - масса или концентрация компонента (масс.% или об.%), принятого за стандарт.
В справочниках приводятся значения калибровочных коэффициентов для многих веществ, и для различных детекторов.
Количественный анализ по методу внутреннего стандарта. Метод предусматривает добавление к известному количеству анализируемой пробы известного количества эталонного соединения (внутреннего стандарта).
В качестве внутреннего стандарта может быть принято как вещество, не содержащееся в пробе, так и содержащееся в ней.
В качестве стандарта используют вещества, удовлетворяющие следующим требованиям: 1) стабильность и инертность, полное смешение с пробой; 2) хорошее разделение со всеми компонентами анализируемой смеси; 3) близость химической природы и параметров удерживания внутреннего стандарта и определяемых соединений; 4) отсутствие примесей. Количество добавки подбирают такое, чтобы площадь пика добавки была соизмерима с площадью пиков компонентов в смеси, подлежащих количественному определению.
Для калибровки следует провести хроматографический анализ ряда смесей вещества-стандарта с каждым из отдельных ожидаемых компонентов при различных соотношениях обоих веществ в смеси.
Если вещество-стандарт не содержится в пробе, то концентрацию определяемого компонента вычисляют по формуле:
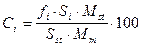
где Ci - концентрация компонента i в анализируемой смеси; Si - площадь его пика; fi - относительный поправочный коэффициент, определяемый по отношению к стандарту; Sst - площадь пика стандарта; Mst - масса добавленного внутреннего стандарта; Mm - масса пробы анализируемой смеси, к которой добавлено определенное количество внутреннего стандарта.
Если вещество-стандарт присутствует в анализируемой пробе, то проводят две серии анализов, в одной из которых анализируют исходную смесь, во второй - исходную смесь с известной добавкой внутреннего стандарта. Количественный анализ методом стандартной добавки проводят по формуле:
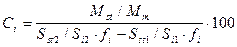
где Ci - концентрация компонента i в анализируемой смеси; Si1 - площадь пика компонента i на хроматограмме исходной смеси; Si2 - площадь пика компонента i на хроматограмме смеси, в которую введен стандарт; fi - относительный поправочный коэффициент, определяемый по отношению к стандарту; Sst1 - площадь пика компонента, принятого за стандарт; Sst2 - площадь пика компонента, принятого за стандарт, на хроматограмме смеси после добавления в нее стандарта, в качестве которого используется компонент, уже содержащийся в смеси; Mst - масса добавленного компонента, принятого за стандарт; Mm - масса пробы анализируемой смеси, к которой добавлено определенное количество стандарта.
Метод внутреннего стандарта можно применять для обработки результатов анализа, выполненного с использованием двух хроматографических колонок.
В качестве стандарта используется компонент, фиксируемый на обоих хроматограммах в виде отдельного пика. Площади (или высоты) пиков на обоих хроматограммах можно сравнивать, воспользовавшись множителем, равным отношению площадей (высот) пика вещества стандарта, зафиксированного на обоих хроматогаммах.
Преимуществами метода является то, что не сказываются отклонения в условиях анализа, нет необходимости в точном дозировании пробы. Недостатком является необходимость специальной подготовки пробы для анализа, трудность в выборе стандарта.
|
|
|
|
|
Дата добавления: 2014-10-31; Просмотров: 5495; Нарушение авторских прав?; Мы поможем в написании вашей работы!