КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Протопорфирии
|
|
|
|
Определение. Протопорфирия (ПрП, эритропоэтическая протопорфирия, эритропеченочная протопорфирия) — заболевание, при котором некоторая светочувствительность кожи сочетается с высокой концентрацией протопорфирина в эритроцитах, обусловленной недостаточностью феррохелатазы. Протопорфирин может накапливаться и в печени.
Генетика, частота и патогенез. Протопорфирия наследуется как аутосомный доминантный признак с непостоянной экспрессивностью. Активность феррохелатазы, катализирующей включение двухвалентного железа в протопорфирин, снижена в костном мозге, периферической крови, печени и культивируемых фибробластах кожи. Эта недостаточность приводит к чрезмерному накоплению протопорфирина в зрелых нормобластах, ретикулоцитах и молодых эритроцитах. По мере старения эритроцитов протопорфирин поступает из них в плазму. Светочувствительность' кожи опосредуется протопорфирином плазмы и кожи и вызывается видимой частью спектра (380—560 нм). Светочувствительность кожи обнаруживает сезонную вариабельность. У некоторых больных печень принимает участие в избыточной продукции порфиринов или, наоборот, может поглощать протопорфирин из плазмы. Многие носители этого дефекта остаются клинически (и химически) здоровыми, и диагноз можно установить только с помощью исследования ферментов.
Клинические проявления и диагностика. Некоторая светочувствительность кожи возникает обычно в детстве. Пребывание на солнце приводит к появлению зуда, эритемы и иногда отека (солнечная крапивница). Через несколько часов или дней эти явления стихают, не оставляя рубцов. Кожные проявления могут возникать лишь после длительного пребывания на солнце. В других случаях начальные изменения кожи прогрессируют до хронической экзематозной фазы (солнечная экзема). При этом заболевании кожа достаточно устойчива к механическим воздействиям, на ней не образуются волдыри в отличие от ПП и ХКП. Не определяются также эритродонтия, гипертрихоз и гиперпигментация. Не наступает и приступов нейропсихических нарушений.
Протопорфирия — болезнь обычно доброкачественная, но может сопровождаться патологией печени, желчных путей или крови. При ней увеличивается частота холелитиаза, причем в состав желчных камней входит протопорфирин. Иногда патология печени вследствие отложения большого количества протопорфирина может прогрессировать до цирроза, приводящего к смерти больного, поэтому у каждого больного следует проводить обычные функциональные печеночные пробы. Нередко протопорфирия сопровождается некоторой анемией.
Диагноз основан на обнаружении высоких концентраций протопорфирина в эритроцитах. При флюоресцентной микроскопии можно видеть большое число флюоресцирующих красным цветом эритроцитов. Уровень протопорфирина может быть повышен и в плазме и кале, тогда как в моче его количество, а также АЛК и ПБГ обычно не изменяется.
Лечение. Местная защита от солнца обычно неэффективна. Пероральный прием b-каротина (обычно в виде смеси р-каротина и кантаксантина) существенно повышает переносимость солнечных лучей. Содержание каротина в сыворотке следует поддерживать на уровне 6000—8000 мкг/л.
ГЛАВА 313. БОЛЕЗНИ НАКОПЛЕНИЯ ГЛИКОГЕНА
Артур Л. Воде (Arthur L. Beaudet)
Болезни накопления гликогена представляют собой группу наследственных нарушений путей накопления углеводов в виде гликогена и путей его утилизации для поддержания уровня сахара в крови и обеспечения тканей энергией. При некоторых формах этой патологии содержание гликогена в тканях не увеличивается.
Гликоген — это сильно разветвленный полимер глюкозы, в котором большинство остатков имеют 1,4-связи, а 7—10% остатков—1,6-связи. Древовидная структура подвергается надстройке и отщеплению остатков на периферии молекулы. Молекулярная масса гликогена составляет несколько миллионов, его молекулы могут агрегировать с образованием структур, видимых при электронной микроскопии. В печени гликогена обычно содержится менее 70 мг/г, а в мышцах — менее 15 мг/г, но эти величины колеблются в зависимости от питания и гормональных влияний. Нарушения структуры гликогена могут быть связаны как с уменьшением, так и с увеличением ветвления молекулы.
Метаболические пути синтеза и распада гликогена схематически изображены на рис. 313-1. В разных тканях эти пути различны; например, некоторые реакции активно протекают в печени, но слабо представлены или отсутствуют в мышцах, а некоторые ферменты в мышцах и печени кодируются разными генами. Глюкоза плазмы проникает в клетку и фосфорилируется глюкокиназой или гексокиназой. Первая содержится в печени, осуществляющей фосфорилирование основной массы глюкозы, тогда как многочисленные гексокиназы распределены по тканям более широко. Глюкозо-6-фосфат (Г-6-Ф) превращается в глюкозо-1-фосфат (Г-1-Ф) в обратимой реакции, катализируемой фосфоглюкомутазой. Уридиндифосфатглюкоза (УФДГ) синтезируется из Г-1-Ф и УТФ под действием УДФГ-пирофосфорилазы. Генетическая недостаточность ни одного из этих печеночных ферментов не была зарегистрирована. Молекула гликогена затем удлиняется путем присоединения отдельных остатков глюкозы из УДФГ, в результате чего образуется полимер. Эта реакция катализируется гликогенсинтазой, которая существует в активной дефосфорилированной и неактивной фосфорилированной формах. Для синтеза нормально разветвленной молекулы гликогена требуется также участие ветвящего (бранчер) фермента (1,4-a-гликан: 1,4-a-гликан-6-гликозилтрансфераза), который переводит 1,4-связи олигосахарида в положение 1,6-связей.
Глюкоза мобилизуется из гликогена целым рядом ферментативных реакций. На гликоген непосредственно действует активная форма фосфорилазы — фосфорилаза а, отщепляя отдельные остатки глюкозы и образуя Г-1-Ф. В мышцах и печени фосфорилаза кодируется разными генными продуктами. В обеих тканях фермент может существовать в активной фосфорилированной и неактивной дефосфорилированной формах. Фосфорилаза представляет собой димер, состоящий из одинаковых субъединиц, причем обе формы фермента подвергаются сложной аллостерической регуляции. Неактивная фосфорилаза b превращается в активную форму под действием фосфорилазо-b-киназы, которая существует и в активной фосфорилированной и неактивной дефосфорилированной формах. Этот фермент состоит из четырех разных субъединиц (a, b, g, d4), причем d-цепь идентична связывающему кальций белку — кальмодулину. Скорость мобилизации глюкозы этой системой регулируется каскадом киназных реакций, включающих цАМФ
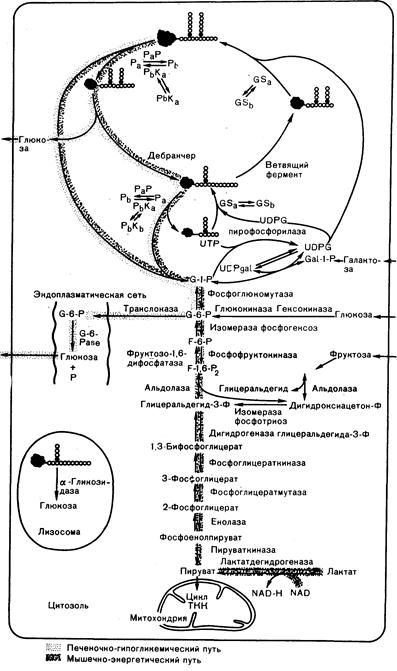
Рис. 313-1. Метаболические пути, имеющие отношение к болезням отложения гликогена.
Изображена гипотетическая клетка, объединяющая особенности печеночной и мышечной. Затененные участки соответствуют реакциям, заблокированным при печеночно-гипогликемических или мышечно-энергетических болезнях. Помимо общепринятых, использованы следующие сокращения: GSa— активная гликогенсинтаза, GSb — неактивная гликогенсинтаза, Ра— активная фосфорилаза, Рb— неактивная фосфорилаза, РаР — фосфатаза фосфорилазы а, РbКа—активная киназа фосфорилазы b, РbКb—неактивная киназа фосфорилазы b.
Таблица 313-1. Болезни накопления гликогена (гликогенозы)
| Тип | Основной дефект' | Клинические проявления | Лабораторные данные | Диагностика | Лечение | Примечание | ||||||||||||
| Печеночно-гипогликемическая патофизиология | ||||||||||||||||||
| 1а (фон Гирке) | Недостаточность глюкозо-б-фосфатазы | Гипогликемия, гепатомегалия, кровоточивость, малый рост, задержка развития, аденомы печени, увеличенные почки | Повышение уровней лактата, холестерина, триглицеридов и мочевой кислоты | Определение ферментов в печени или кишечнике, увеличение запасов гликогена с нормальной структурой в печени | Частый прием пищи, ночное кормление через зонд, 60—70 % углеводов, ограничение сахарозы и лактозы, при необходимости гидрокарбонат и аллопуринол | Частое тяжелое аутосомно-рецессивное заболевание | ||||||||||||
| Недостаточность микросомальной Г-б-Ф-транслоказы | То же, что при 1а, а также нейтропения и повторные рецидивирующие инфекции | То же | Определение ферментов в печени с детергентом или без него | То же | Редкое тяжелое аутосомно-рецессивное заболевание | |||||||||||||
| III (Кори) | Недостаточность дебран-чера | Гипогликемия, гепатомегалия, у некоторых больных малый рост и задержка развития, легкая миопатия, утяжеляющаяся у некоторых взрослых больных | Уровни лактата и мочевой кислоты в пределах нормы, повышение уровней холестерина, триглицеридов и глу-тамикооксалаце-таттрансаминазы в сыворотке | Определение ферментов в печени мышцах или фибробластах; непостоянные изменения в лейкоцитах; увеличение уровня гликогена с измененной структурой в печени и мышцах | Частый прием пищи, ночное кормление через зонд, 50 % углеводов и 15—20 % белка | Частое, умеренной тяжести заболевание; умеренный фиброз печени | ||||||||||||
| VI (Эра) | Недостаточность фосфорилазы в печени | Гепатомегалия, непостоянная гипогликемия | Минимальные изменения,? гиперлипидемия | Определение ферментов в печени; повышенный уровень гликогена с нормальной структурой в печени | Диета та же, что при типе III, серьезного лечения часто не требуется | Редкое и недостаточно охарактеризованное заболевание;? аутосомно-рецессивное наследование | ||||||||||||
| Патология, ранее называемая VI6, VIII или IX | Недостаточность фосфо-рилазо-Ь-кина-зы в печени | Гепатомегалия, непостоянная гипогликемия, случайные находки у женщин-гетерозигот | Минимальные изменения | Определение ферментов в лейкоцитах, фибробластах, или печени; повышенный уровень гликогена с нормальной структурой в печени | То же | Очень легкое, но, возможно, широко распространенное заболевание, сцепленное с Х-хромосомой | ||||||||||||
| Мышечно-энергетическая патофизиология | ||||||||||||||||||
| V (Мак-Ардл) | Недостаточность фосфорилазы в мышцах | Боли, судороги и миоглобинурия при тяжелой физической нагрузке | В периоды приступов повышен уровень КФК, недостаточная продукция лактата при тесте с ишемической нагрузкой | Определение ферментов в мышцах, повышенный уровень гликогена с нормальной структурой в мышцах | Исключение нагрузок, прием глюкозы или фруктозы перед нагрузкой | В некоторых случаях явное аутосомно-рецессивное наследование, болеют в основном мужчины | ||||||||||||
| VII | Недостаточность фосфофруктокиназы в мышцах | То же, что при типе V, а также незначительная гемолитическая анемия | То же | То же | То же | Редкое заболевание, аутосомно-рецессивное наследование | ||||||||||||
| Недостаточность фосфо-глицеромутазы в мышцах | То же, что при типе V | »» | Определение ферментов в мышцах, нормальное содержание гликогена | ? То же | Выявлен всего один больной (мужчина) | |||||||||||||
| Недостаточность М-субъ-единицы ЛДГ | То же | В периоды приступов повышен уровень КФК; при тесте с ишемической нагрузкой повышается уровень пирувата, но не лактата | Определение изоферментов ЛДГ в сыворотке, эритроцитах или лейкоцитах; определение ферментов в мышцах;? нормальное содержание гликогена | ? То же | Выявлено трое больных мужчин и одна женщина | |||||||||||||
| Индивидуальные особенности патофизиологии | ||||||||||||||||||
| II (Помпе) | Недостаточность лизосомной а-глюко-зидазы | Инфантильная форма: гипотензия, мышечная слабость, увеличение размеров сердца и сердечная недостаточность, увеличение размеров языка, ранняя смерть; ювенильная форма: прогрессирующая слабость скелетных | Повышенная КФК без гипогликемии | Определение ферментов в мышцах или фибробластах, возможно определение ферментов в лейкоцитах (чревато серьезными ошибками) | Эффективного лечения не разработано | Частое, аутосомно-рецессивное заболевание; возможен пренатальный диагноз; проводится широкое обследование новорожденных | ||||||||||||
| мышц; взрослая форма: прогрессирующая слабость скелетных мышц явления легочной недостаточности | , | |||||||||||||||||
| IV (Андерсена) | Недостаточность ветвя-щего фермента | Замедленное развитие новорожденных, цирроз и недостаточность печени, резкая гипотензия и слабость у некоторых больных, ранняя смерть | Уровень сахара в пределах нормы, изменения, характерные для болезни печени | Определение ферментов в печени, мышцах, лейкоцитах или фибробластах; уровень гликогена изменен слабо, но его структура нарушена | То же | Очень редкое заболевание с аутосомно-рецессивным типом наследования | ||||||||||||
Эти нарушения определяют предпочтительное название болезни.
К мышечно-энергетическим нарушениям относятся недостаточность мышечной фосфорилазы (тип V), фосфофруктокиназы (тип VII), фосфоглицеромутазы и М-субъединицы ЛДГ. В клинической картине при этой патологии доминируют боли в мышцах, миоглобинурия и повышение уровня мышечных ферментов в сыворотке при тяжелой физической нагрузке. Все эти состояния объединяют разрыв цепи реакций от гликогена до лактата с сопутствующей неспособностью окислять НАД-Н. Неувеличивающийся уровень лактата в крови в ответ на физическую нагрузку служит диагностическим признаком нарушения мышечно-энергетического типа. Недостаточность блокирующего ветвление фермента формирует смешанный синдром; в основном эта недостаточность проявляется как нарушение печеночио-гипогликемического типа, и количества глюкозы, высвобождаемой под действием фосфорилазы, по-видимому, достаточно для предотвращения миоглобинурии, но не миопатии и слабости скелетных мышц.
Два других нарушения следует обсуждать каждое в отдельности. Недостаточность лизосомной a-глюкозидазы — это лизосомная патология, не оказывающая сильного влияния ни на углеводный обмен, ни на механизмы поддержания уровня сахара в крови (см. гл. 316). Основной патологический процесс при недостаточности ветвящего фермента — это выраженный цирроз печени, вероятно, вследствие повреждающего эффекта накопления гликогена с нарушенной структурой. Количество гликогена обычно не изменено. Не нарушается и способность поддерживать нормальный уровень сахара в крови.
|
|
|
|
|
Дата добавления: 2014-11-20; Просмотров: 485; Нарушение авторских прав?; Мы поможем в написании вашей работы!