КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Болезни с индивидуальной патофизиологией
|
|
|
|
Недостаточность a-глюкозидазы, тип II. Этот тип недостаточности, или болезнь Помпе, представляет собой лизосомную болезнь накопления, патофизиология которой обсуждается в гл. 316. Частота ее неизвестна, но может превышать 1:100000. Она не сопровождается гипогликемией, кетозом или другими нарушениями межуточного обмена.
Инфантильная форма болезни проявляется в первые 6 мес жизни, а иногда сразу после рождения. Клиника заключается в гипотензии и слабости скелетных мышц, значительном увеличении размеров сердца, увеличении размеров языка и разной степени гепатомегалии. Уровень мышечных ферментов, например креатинфосфокиназы и альдолазы в сыворотке, обычно повышен, а на ЭКГ можно видеть увеличенные комплексы QRS с укорочением интервала Р — R. Иногда появляются двигательные нарушения и задержка развития. В большинстве случаев больной умирает в возрасте 2—3 лет от сердечной недостаточности.
Ювенильная форма болезни характеризуется признаками прогрессирующей мышечной дистрофии. В этом случае нарушается походка, но сердечные проявления отсутствуют. Уровень креатинфосфокиназы и альдолазы в плазме повышен. Продолжительность жизни варьирует. Еще более легкая взрослая форма проявляется слабостью скелетной мускулатуры в возрасте 20—50 лет. Изменения со стороны сердца опять-таки отсутствуют, уровень мышечных ферментов в сыворотке повышен. У некоторых больных развивается дыхательная недостаточность из-за вовлечения в процесс дыхательной мускулатуры, поэтому им часто ошибочно ставят диагноз одной из форм мышечной дистрофии.
В биоптатах мышц обнаруживают вакуолизацию клеток и увеличение количества гликогена. При электронной микроскопии можно видеть связанные с мембраной вакуоли, содержащие гликоген, что служит диагностическим признаком. Избыток гликогена находят и в других тканях, в том числе в печени и центральной нервной системе, особенно в клетках передних рогов спинного мозга. Окончательный диагноз устанавливают с помощью определения ферментов в биоптатах мышц, печени или в культуре фибробластов. Как правило, у больных со взрослой формой болезни сохраняется некоторая остаточная активность фермента, но ее точная величина не имеет прогностического значения. Возможна пренатальная диагностика, позволяющая выявить инфантильную форму болезни. Предпринимались попытки использования разных видов ферментной инфузионной терапии, но все они не увенчались успехом.
Недостаточность ветвящего фермента, тип IV. Этот тип недостаточности, или болезнь Андерсена, представляет собой редкое заболевание с аутосомным рецессивным наследованием. Ее признаки у новорожденных заключаются в гепатомегалии, замедлении развития и гипотензии в первые месяцы жизни с последующим прогрессирующим циррозом печени. У других больных ведущими признаками служат изменения сердца и/или резчайшая гипотензия, сходная с таковой при спинальной мышечной атрофии и дегенерации клеток передних рогов. Смерть наступает в первые 2—3 года жизни.
Полагают, что симптоматика связана в первую очередь с нарушением структуры гликогена вследствие генерализованной недостаточности ветвящего фермента. Длинные наружные цепи в молекулах гликогена обусловили название болезни «амилопектиноз». Данные лабораторных исследований обычно типичны для выраженного повреждения печени, за тем исключением, что гипогликемия, как правило, не развивается. Отсутствие гипогликемии и нормальное количество гликогена в печени затрудняют диагностику. Болезнь предполагают при выявлении нарушенной структуры гликогена в биоптатах, и диагноз подтверждают путем непосредственного определения фермента в печени, лейкоцитах или культуре фибробластов кожи. Эффективного лечения не разработано. Диагноз можно установить пренатально, используя клетки амниотической жидкости.
Другие возможные нарушения метаболизма гликогена. Сообщалось о случаях недостаточности гликогенсинтазы у некоторых больных. Обычно у них определяют гипогликемию натощак, судороги и некоторые нарушения психики. Сохранение какого-то количества гликогена в печени, повышение уровня глюкозы в плазме в ответ на введение глюкагона или галактозы и известная лабильность системы активации гликогенсинтазы породили скептическое отношение по поводу существования этой аномалии. Этот синдром можно спутать с кетозной гипогликемией у детей (см. гл. 329).
Сообщается также о нескольких ферментных дефектах у одного и того же больного и разных ферментных дефектах у сиблингов. Многие из этих сообщений могут быть результатом методических трудностей определения ферментов в измененных тканях человека. В настоящее время какие-либо специфические синдромы множественной первичной недостаточности ферментов не доказаны.
ГЛАВА 314. ГАЛАКТОЗЕМИЯ, НЕДОСТАТОЧНОСТЬ ГАЛАКТОКИНАЗЫ И ДРУГИЕ РЕДКИЕ НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА
Курт Дж. Иссельбахер (Kurt J. Isselbacher)
Определение. Термином «галактоземия» обозначают два вида врожденных нарушений обмена галактозы: классическая галактоземия обусловливается недостаточностью галактозо-1-фосфатуридилтрансферазы (ГАЛТ) и сопровождается катарактой, умственной отсталостью и циррозом печени, а недостаточность галактокиназы обусловливает главным образом образование катаракты.
Патогенез. Лактоза — основной углевод молока — представляет собой дисахарид, содержащий галактозу и глюкозу. В пищеварительных путях она гидролизуется кишечной лактазой. В норме всосавшаяся галактоза в печени превращается в глюкозу. Первой реакцией на этом пути является фосфорилирование галактозы с образованием галактозо-1-фосфата под действием галактокиназы, кодируемой геном, расположенным на 17-й хромосоме:

На следующем этапе галактозо-1-фосфат превращается в глюкозо-1-фосфат под действием ГАЛТ, ген которой расположен на 9-й хромосоме:
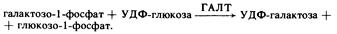
УДФ-сахара могут превращаться один в другой в процессе эпимеразной реакции:
УДФ-галактоза Û УДФ-глюкоза.
Галактоза метаболизируется и другими путями. В присутствии НАДФ•Н (или НАД-Н) она может превращаться (восстанавливаться) в галактитол (дульцитол) под действием альдозоредуктазы. Небольшое ее количество может и окисляться галактозодегидрогеназой с образованием галактоновой кислоты, ксиулозы и двуокиси углерода. Эти пути и определяют ограниченный метаболизм галактозы у больных с галактоземией.
При недостаточности галактокиназы галактоза накапливается в крови и тканях. В хрусталике глаза она под действием альдозоредуктазы превращается в галактитол — сахар, для которого хрусталик непроницаем. В результате происходит чрезмерная гидратация, которая на фоне уменьшения количества глутатиона в хрусталике обусловливает развитие катаракты.
При классической галактоземии недостаточность ГАЛТ приводит к накоплению в тканях галактозо-1-фосфата и галактозы. Как и при недостаточности галактокиназы, катаракта развивается вследствие накопления в хрусталике галактитола. Предполагают, что цирроз печени и задержка психического развития связаны с увеличением количества галактозо-1-фосфата в этих тканях. Повышенный уровень галактозы в крови может снижать печеночную продукцию глюкозы, обусловливая гипогликемию. В почках и кишечнике накопление галактозы и галактозо-1 -фосфата приводит, по-видимому, к торможению транспорта аминокислот. У некоторых женщин нарушается функция яичников в связи с гипергонадотропным гипогонадизмом, патогенез которого неизвестен.
Как недостаточность галактокиназы, так и недостаточность ГАЛТ наследуются по аутосомному рецессивному типу. У гетерозигот по этим нарушениям уровни ферментов составляют лишь половину от нормы, но симптоматика болезни отсутствует. Недостаточность галактокиназы у беременной, потребляющей лактозу, может обусловить развитие катаракты у плода. Однако не все больные с недостаточностью ГАЛТ в клетках бывают носителями классической галактоземии. У некоторых лиц, гомозиготных по другому гену, называемому вариантом Дуарте, уровень ГАЛТ обычно также в 2 раза ниже нормы, но симптоматика при этом отсутствует. Их можно отличить от больных с классической галактоземией по данным исследования электрофоретических свойств мутантного фермента. Как при недостаточности галактокиназы, так и при классической галактоземии может быть выявлена либо функциональная недостаточность, либо отсутствие необходимого фермента. Классическая галактоземия обусловливается мутацией структурного гена, поэтому измененный фермент (ГАЛТ) лишен нормальной функции. Известны и другие клинические варианты с измененной электрофоретической подвижностью фермента.
Частота классической галактоземии среди представителей европеоидной популяции составляет примерно 1:80000 новорожденных. Около 0,8—1,3 % населения представляют собой гетерозиготы по гену галактоземии (ГАЛТ) и примерно 10 % — носители варианта Дуарте. При скрининге новорожденных на галактоземию наиболее частой причиной патологии служит компаунд-гетерозиготность по варианту Дуарте и классической галактоземии, при которой содержание ГАЛТ находится на уровне 17 % от нормы. Клинические проявления у них отсутствуют.
Клинические проявления. Симптомы классической галактоземии обычно появляются в первые несколько дней или недель после рождения. Ребенок отказывается от грудного молока или молочных продуктов, у него появляются рвота и признаки недоедания, его развитие замедляется. Могут присоединиться желтуха, гепатомегалия и признаки печеночной патологии. Обычно у новорожденных катаракта не выявляется, но развивается через несколько недель или месяцев. Отсталость психического развития становится очевидной в возрасте 6—12 мес. У детей с классической галактоземией часто развивается бактериальный сепсис (особенно обусловленный кишечной палочкой), который может быть основной причиной смерти новорожденного. Единственным постоянным симптомом недостаточности галактокиназы служит катаракта.
Диагностика. Недостаточность галактокиназы следует подозревать у младенцев или детей с катарактой, в моче которых определяют отличающиеся от глюкозы восстанавливающие вещества. Диагноз устанавливают при выявлении дефицита галактокиназы в эритроцитах.
О классической галактоземии следует думать, если налицо один из упомянутых признаков или более. При потреблении молока в моче больного появляется восстанавливающий сахар, неопределяемый с помощью глюкозоксидазной реакции (т. е. не глюкоза), но идентифицируемый как галактоза с помощью других (например, хроматографических) методов. Если у ребенка появляется рвота, снижается аппетит или ему вводят глюкозу внутривенно, то галактоза в моче может и не определяться. Окончательный диагноз устанавливают, убедившись в недостаточности или отсутствии ГАЛТ в эритроцитах. Для этого используют разные методы. Заболевание можно диагностировать и пренатально, исследуя ферменты в культуре клеток, полученных при амниоцентезе, или обнаружив увеличенное количество галактитола в амниотической жидкости.
В неонатальном периоде галактоземию следует дифференцировать от первичной печеночной патологии. При болезни печени нарушается извлечение галактозы из крови и может повышаться уровень галактозы в крови и в моче. Однако в этих случаях содержание ГАЛТ остается в пределах нормы.
Лечение. Оно заключается в исключении из диеты продуктов, содержащих галактозу, особенно молока. Часто используют заменители молока, такие как нутрамиген. Несмотря на то что препараты из бобов сои содержат галактозу в составе полисахаридов, они, по-видимому, хорошо переносятся больными, так как связанная галактоза высвобождается очень медленно. У больных детей, вскармливаемых препаратами сои, уровень галактозо-1-фосфата в эритроцитах, как правило, не повышается.
Перевод больного на безгалактозную диету обычно сопровождается резким ослаблением симптоматики, за исключением задержки психического развития. Больного следует содержать на безгалактозной диете в течение неопределенно продолжительного периода или, по крайней мере, до тех пор, пока не нормализуется их физическое и неврологическое состояние.
Другие нарушения углеводного обмена. Признаки наследственного нарушения толерантности к фруктозе и недостаточности фруктозо-1,6-дифосфатазы — двух видов аутосомных рецессивных нарушений метаболизма фруктозы, приводящих к гипогликемии, суммированы в табл. 314-1 (см. также гл. 313 и 329).
Таблица 314-1. Некоторые другие нарушения углеводного обмена
| Нарушение | Метаболический дефект | Проявления |
| Наследственное нарушение толерантности к фруктозе | Недостаточность фруктозо-1-фосфатальдолазы приводит к накоплению фруктозо-1-РО4 в тканях | Печеночная патология, повреждение почечных канальцев и гипогликемия |
| Недостаточность фруктозо-1,6-дифосфатазы | Недостаточность фермента блокирует глюконеогенез из обычных предшественников — лактата, глицерина и аланина, поэтому уровень глюкозы в крови зависит от поступления экзогенной глюкозы | Лактацидоз приводит к гипервентиляции, сонливости и коме обычно на фоне гипогликемии и кетоза |
ГЛАВА 315. ГИПЕРЛИПОПРОТЕИНЕМИИ И ДРУГИЕ НАРУШЕНИЯ ЛИПИДНОГО ОБМЕНА
Майкл Е. Браун, Джозеф Л. Гольдштейн (Michael S. Brown, Joseph L. Goldstein)
Гиперлипопротеинемии представляют собой нарушения транспорта липидов, обусловленные ускоренным синтезом или замедленным разрушением липопротеинов, переносящих холестерин и триглицериды в плазме. Повышение уровня липопротеинов в плазме имеет важное клинические значение потому, что они могут обусловливать развитие двух тяжелых, угрожающих жизни заболеваний — атеросклероза и панкреатита. Уменьшение количества содержащегося в липопротеинах холестерина, осуществляемое с помощью диеты и лекарственных средств, уменьшает при гиперлипопротеинемии риск инфаркта миокарда. Одни гиперлипопротеинемии обусловливаются непосредственно первичным нарушением процессов синтеза и разрушения липопротеиновых частиц. Другие развиваются вторично, т. е. повышение уровня липопротеинов в плазме служит одним из проявлений аномалий, связанных с нарушением регуляторных метаболических систем, например с недостаточностью тиреоидных гормонов или инсулина. Первичные гиперлипопротеинемии можно разделить на две большие группы: 1) нарушения одиночного гена, которые передаются простым доминантным или рецессивным механизмом; 2) многофакторные нарушения со сложным характером наследования, при которых гиперлипопротеинемии разной тяжести у членов одной семьи обусловливаются взаимодействием слабых эффектов многочисленных вариантных генов с эффектами факторов внешней среды.
|
|
|
|
|
Дата добавления: 2014-11-20; Просмотров: 469; Нарушение авторских прав?; Мы поможем в написании вашей работы!