КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Металлические электроды
|
|
|
|
Скорость гетерогенных реакций измеряется количеством вещества, вступающего в реакцию или образующегося в единицу времени на единицу поверхности. Процесс осуществляется не в объеме твердого вещества, а на поверхности, концентрация его остается постоянной, поэтому скорость гетерогенных реакций определяется изменением концентрации газов или жидкостей.
Скорость реакции зависит от природы реагирующих веществ, их концентрации, температуры, давления (для реакций с участием газов), присутствия
в системе катализаторов, от поверхности взаимодействия реагирующих веществ (в случае гетерогенной реакции) и т.д.
В зависмости от числа фаз сист. бывают: гомогенные(1фаза); гетерогенные(2 и более фаз).
Ср. скорость р-ции-изм. кол-ва в-ва в еденицу времени в еденице реакц. пр-ва
Если Δt стремится к 0, то в пределе ср. скорость становится скоростью р-ции в данный момент времени или мгновенной скоростью.
Практ. оценка V (относительная)=1/t.
Гетерогенные реакции представ. собой сложные взаимод. и в общем случае вкл. след. стадии: 1.проц. переноса (диффузия)исходных в-в и пов. раздела фаз; 2.адсорбция-.осаждение реаг. в-в на пов-ти; 3.химическая р-ция; 4.десорбция-удаление продуктов р-ции от границ раздела фаз;5.диффузия продуктов р-ции по всему объему;
Скорость р-ции завист:от природы реаг. в-в.; условий протекания р-ции(C,T,P);
присутсвие катализаторов; величины реагир. поверхности.(для гетерог. р-ций).
3. Зависимость V х. р. От концентрации. Закон действующих масс (ЗДМ) для гомогенных и гетерогенных х. р. (на конкретных примерах). Константа скорости и её физический смысл. От каких факторов зависит? (стр.6)
Зависимость скорости реакции от концентрации реагирующих веществ выражается основным законом химической кинетики – законом действия масс (ЗДМ): скорость гомогенной химической реакции при постоянной температуре прямо пропорциональна произведению концентраций реагирующих веществ, взятых в степенях, равных стехиометрическим коэффициентам в уравнении реакции.
Для реакции 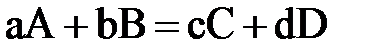 выражение закона запишется как
выражение закона запишется как
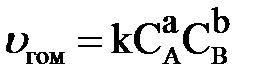 , ,
| (1.1) |
где 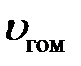 – скорость реакции;
– скорость реакции;  – константа скорости химической реакции;
– константа скорости химической реакции;
СА и СВ – концентрации реагирующих веществ, моль/л; а, b – стехиометрические коэффициенты в уравнении реакции.
Физический смысл константы скорости (k): k показывает, с какой скоростью протекает реакция при концентрациях реагирующих веществ, равных 1 моль/л.
Константа скорости зависит от природы реагирующих веществ, температуры, присутствия катализатора, но не зависит от концентрации реагирующих веществ и парциальных давлений (для газов).
В случае гетерогенных процессов в закон действия масс входят концентрации только тех веществ, которые находятся в газовой фазе или растворе. Концентрации веществ, находящихся в твердой фазе, постоянны и включены в константу скорости.
Пример: 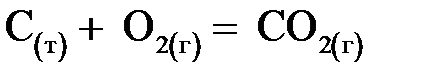 ;
; 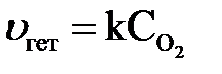 или
или 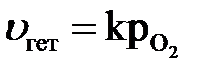 ;
;
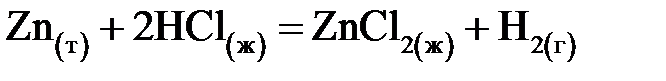 ;
; 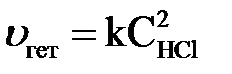 .
.
В общем случае скорость реакции в гетерогенных процессах пропорциональна поверхности соприкосновения реагирующих веществ. Чем больше поверхность взаимодействия, тем больше вероятность столкновения взаимодействующих частиц, и, следовательно, больше скорость гетерогенной реакции.
Закон действия масс справедлив для простых реакций, протекающих в газах или растворах.
4. Зависимость V х. р. от температуры: правило Вант-Гоффа, уравнение Аррениуса. (стр.7)
Зависимость скорости физико-химического процесса от температуры приближенно выражается правилом Вант-Гоффа: при увеличении температуры на каждые 10 градусов скорость большинства химических реакций возрастает примерно в 2 –4 раза. Математически эта зависимость выражается соотношением
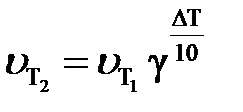 или или 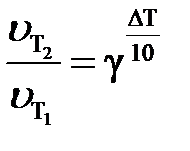 , ,
| (1.3) |
где 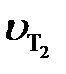 и
и 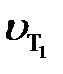 – cкорости реакции при температурах Т2 и Т1; ∆Т = Т2 – Т1;
– cкорости реакции при температурах Т2 и Т1; ∆Т = Т2 – Т1;
γ – температурный коэффициент скорости (значения изменяются от 2 до 4), показывающий, во сколько раз увеличится скорость реакции с повышением температуры на 10 градусов. Численное значение γ зависит от природы реагирующих веществ и для данной реакции есть величина постоянная.
Функциональную зависимость скорости реакции от температуры выражает уравнение Аррениуса:
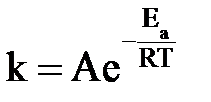 или или 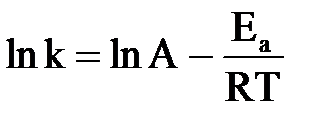 , ,
| (1.4) |
где k – константа скорости; А – постоянная величина для реакции, характеризует общее число столкновений с благоприятной ориентацией, не зависит от температуры; е – основание натурального логарифма; Еа – энергия активации, 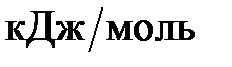 ; R – универсальная газовая постоянная; Т – абсолютная температура, К.
; R – универсальная газовая постоянная; Т – абсолютная температура, К.
Множитель е– Еа / RT, называемый экспоненциальным, характеризует долю активных столкновений (столкновений активных частиц) от их общего числа. Анализ уравнения Аррениуса показывает: при повышении температуры возрастает доля активных столкновений, объясняя экспоненциальную зависимость скорости реакции от температуры.
Следствие из уравнения Аррениуса (1.4): при увеличении температуры в большей степени растет константа скорости той реакции, энергия активации которой больше.
Зная 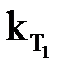 и
и 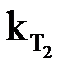 , можно вычислить Еа из соотношения
, можно вычислить Еа из соотношения
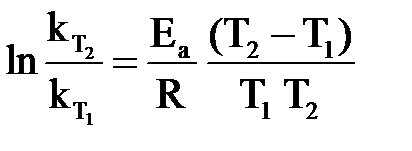 или или 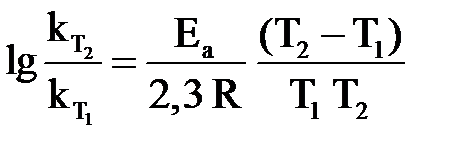 . .
| (1.5) |
Одним из наиболее распространенных в практике способов изменения скорости процессов является катализ. При катализе реакция протекает через ряд промежуточных стадий, каждая из которых характеризуется определенным значением энергии активации: катализаторы уменьшают Еа, ингибиторы (замедлители) увеличивают Еа.
5. Теория активированного комплекса. Энергия активации, её физический смысл. Энергетическая диаграмма экзо- и эндотермических обратимых реакций. (стр.7)
Увеличение скорости химической реакции с повышением температуры связано с возрастанием числа активных молекул, обладающих избыточной энергией. Избыточная энергия, которой должны обладать молекулы, чтобы столкновение было эффективным, называется энергией активации Еа. Численное значение Еа зависит от природы реагирующих веществ и катализатора. Чем больше значение Еа, тем меньше скорость химической реакции.
| Энергетические диаграммы реакций- описаниехода реакции через изменение энергии исходных веществ. Величина Еа опред. высоту энергет. барьера реакции.Преодоление этого барьера частицами исх. в-в связано с обр. активного комплекса. Активный комплекс -группа взаим. частиц. в момент соударения: старые связи ослаблены, новые наметились, но ещё не образов. Ослабление старых связей через переходн. сост.-необходимое усл. перегруппировки атомов и образов. новых молекул. В общем случае активация частиц достигается: увеличением температуры; действием электронного поля; облучением и т.д. В газовых реакциях основн. источником активации явл. соударение частицами обл. большим запасом энергии. |
Есть две возможности ускор. хим реакцию: увеличение темп; снижение энергии активации с помощ. катализатора.
| Введение в сист. катализатора разбивает проц. на ряд промежут. стадий, энергия активации кот. меньше Еа самого проц. Действие катализаторов: 1.не изменяет теплового эффекта р-ции. 2.снижает энергию активации как прямой так и обратной р-ции на одну и туже величину, поэтому не смещает равновесия. 3.увеличивая в одинаковой степени скорость прямой и обратной р-ции, сокращает время достижения равновесия. 4.является избирательным,что определяется природой катализатора и условиями его применения. |
6. Изменение V пр. и V обр. химических реакций во времени. Кинетические условия наступления равновесия. Вывод константы равновесия Кс и Кр и их взаимосвязь. (стр.8)
По принципу обратимости реакции можно разделить на обратимые, идущие одновременно в двух противоположных направлениях, и необратимые, идущие до конца в данном направлении. Обратимые реакции в закрытой системе при постоянной температуре и давлении идут до состояния равновесия.
Химическое равновесие – состояние обратимого процесса, при котором скорости прямой и обратной реакции равны. Концентрации реагирующих веществ, установившиеся к моменту наступления равновесия, называют равновесными, они остаются постоянными до нарушения химического равновесия.
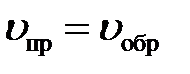 – кинетическое условие равновесия.
– кинетическое условие равновесия.
Численно химическое равновесие характеризуется величиной константы равновесия. В общем случае для обратимой реакции 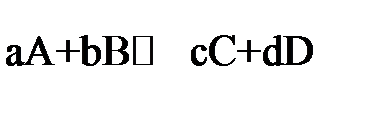
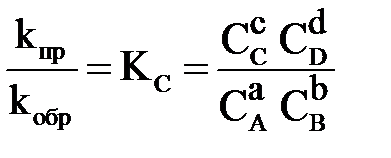 , ,
| (1.6) |
где КС – константа равновесия; kпр, kобр – константы скорости прямой и обратной реакции; СС, СD, CA, CB – равновесные концентрации веществ, моль/л; а, b, с, d – cтехиометрические коэффициенты в уравнении реакции.
Для газообразных систем можно использовать равновесные парциальные давления газов. Тогда
 . .
| (1.7) |
В гетерогенных системах в выражения константы равновесия не входят концентрации твердых веществ. Например, для равновесной системы
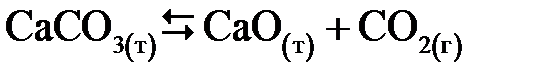 ,
, 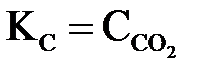 или
или 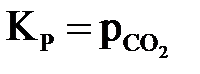 .
.
7. Физический смысл константы равновесия. Зависимость от температуры, уравнение изобары. (стр.9)
Физический смысл КС ( КР ): константа равновесия определяет глубину протекания процесса к моменту достижения системой равновесного состояния. Чем больше численное значение К, тем больше степень превращения исходных веществ в продукты реакции и, следовательно, тем c большей скоростью до достижения равновесия идет прямая реакция.
Константа равновесия зависит от температуры и природы реагирующих веществ и не зависит от концентраций (парциальных давлений), присутствия катализатора. Введение катализатора в систему не влияет на отношение kпр / kобр, т.е. на значение константы равновесия: одинаково уменьшается энергия активации и увеличивается константа скорости как прямой, так и обратной реакций.
Увеличение или уменьшение константы равновесия при изменении температуры определяется соотношением между энергиями активации прямой и обратной реакций. Разность энергий активации прямой и обратной реакций определяет тепловой эффект процесса | DН | = | Еа. пр – Еа. обр|.
8. Химическое равновесие и его динамичность. Условия сохранения и смещения равновесия. Принцип Ле Шателье. (стр.10)
Состояние химического равновесия существует при строго определенных условиях: концентрации, температуре, давлении. При изменении одного из этих условий равновесие нарушается вследствие неодинакового изменения скоростей прямой и обратной реакций. Переход из одного равновесного состояния в другое называется сдвигом или смещением положения равновесия. Если скорость прямой реакции становится больше скорости обратной реакции, равновесие смещается вправо. Если скорость прямой реакции становится меньше, чем скорость обратной, то равновесие смещается влево. С течением времени
в системе устанавливается новое химическое равновесие, которое характеризуется равенством скоростей 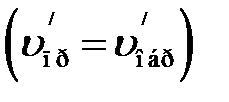 и новыми равновесными концентрациями всех веществ в системе.
и новыми равновесными концентрациями всех веществ в системе.
Направление смещения равновесия определяется принципом Ле Шателье: если на систему, находящуюся в равновесии, оказывается внешнее воздействие, то равновесие смещается в направлении той реакции, которая ослабляет это воздействие.
Кинетич. условие равновес. Vпр=Vобр.
Признаки хим. равновес:
1.Хим. равновес. не зависит от пути его достижения.
2.Сост. равновесия не изм. при пост. Р,Т,С
3.Явл. динамичным, т.е.изменяется при изменении Р,Т,С.
4.Концентрация в-в или парцианально давление газов, устанав.к моменту равновес., наз. равновесным, они ост. постоян. до нарушения хим. равновес.
Состояние хим равновесия существует при строго определённых условиях:С.P.T.при изменении одного из этих условий-равновесие нарушается.
Смещение хим равновесия-переход системы из одного равновесного состояния в другое, вызванный неодинаковым изменением Vпр и Vобр, вследствии изменения одного из условий при котором система находилась в равновесии.
Если Vпр>Vобр то равн смещ. вправо
Если Vпр<Vобр то равн смещ. влево
Напрвление смещения равновесия определятся принципом Ле Шателье:Если на систему находящуюся в равновесии оказывается внешнее воздействие, то равновесие смещается в направлении той р-ции, которая ослабляет это воздействие.
9. Расчет эквивалентных масс простых и сложных веществ. Эквивалентные объемы и их расчет для Н2, О2, Cl2.
10. Растворы и способы выражения их концентраций: молярность и нормальность и их взаимосвязь.
11.Растворы электролитов и их классификация: кислоты, основания (гидроксиды), соли. Примеры их диссоциации.
Электролиты – вещества, которые в растворах и расплавах полностью или частично состоят из ионов и проводят электрический ток. К электролитам относятся водные растворы кислот, оснований и солей, а также расплавы многих солей, оснований, оксидов, гидридов s-металлов.
Электропроводность электролитов обусловлена направленным движением ионов в электрическом поле. Растворы (расплавы) электролитов являются ионными проводниками, или проводниками второго рода. (Проводниками первого рода называют проводники с электронным типом проводимости).
Электролитическая диссоциация – процесс распада молекул электролита на ионы. В растворах диссоциация происходит под действием молекул растворителя.
Электролитической диссоциации подвергаются ионные соединения и молекулярные соединения с полярной ковалентной связью в полярных растворителях, среди которых важнейшим является вода. При электролитической диссоциации в воде ионы гидратированы, т.е. окружены оболочкой из молекул воды, свободные ионы в водных растворах отсутствуют. Уравнения электролитической диссоциации обычно записываются в упрощенной форме без указания гидратной оболочки.
Электролиты при растворении в воде распадаются (диссоциируют) на ионы: положительно заряженные – катионы (Na+, Al3+, Cu2+) и отрицательно заряженные – анионы (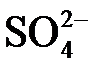 ,
, 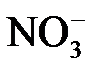 ,
, 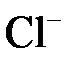 ). При этом раствор остается в целом электронейтральным: алгебраическая сумма зарядов ионов равняется нулю.
). При этом раствор остается в целом электронейтральным: алгебраическая сумма зарядов ионов равняется нулю.
С точки зрения теории электролитической диссоциации кислотами называются вещества, которые диссоциируют в воде на ионы водорода 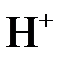 , основаниями – вещества, диссоциирующие на гидроксид-ионы
, основаниями – вещества, диссоциирующие на гидроксид-ионы 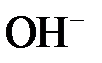 , солями – соединения, образующие при диссоциации катионы металлов и анионы кислотных остатков.
, солями – соединения, образующие при диссоциации катионы металлов и анионы кислотных остатков.
12.Сильные и слабые электролиты. Степень и константа диссоциации и их взаимосвязь. Расчет молярных концентраций ионов.(стр.18)
Степень диссоциации. Сильные и слабые электролиты
Количественно процесс диссоциации электролитов характеризуется степенью диссоциации (a). Степень диссоциации – это отношение числа молекул, распавшихся на ионы (N), к общему числу молекул электролита (N0):
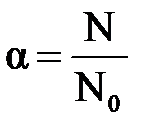 . .
| (2.6) |
Степень диссоциации показывает долю молекул вещества, распавшихся на ионы и, таким образом, характеризует силу электролита. Электрическая проводимость растворов электролитов тем выше, чем больше ионов в растворе, т.е. больше степень диссоциации.
Численное значение aзависит от природы растворенного вещества, температуры, концентрации раствора и принимает значение, меньше или равное единице. По значению a все электролиты условно делят на сильные ( a ® 1) и слабые ( a << 1).
Cильными электролитами являются хорошо растворимые соли; кислоты: галогеноводородные – (кроме HF), H2SO4, HNO3, HClO3, HClO4, HMnO4; основания щелочных и щелочно-земельных металлов – NaOH, KOH, RbOH, CsOH, Ca(OH)2, Sr(OH)2, Ba(OH)2.
Слабыми электролитами – почти все карбоновые кислоты; некоторые минеральные кислоты (H2CO3, H2SO3, H2S, HCN, HF, HClO, HNO2, и др.); многие основания (Cu(OH)2, Fe(OH)2, Ni(OH)2 и др.); малорастворимые соли (BaSO4, CaSO4, AgCl); гидроксид аммония (NH4OH) и вода.
Концентрацию ионов 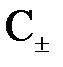 (катионов и анионов) в растворе сильного электролита можно рассчитать по формуле
(катионов и анионов) в растворе сильного электролита можно рассчитать по формуле
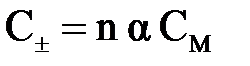 , ,
| (2.7) |
где n – число ионов данного вида, на которое распадается одна молекула вещества при диссоциации; α – степень диссоциации; CM – молярная концентрация раствора, моль/л.
Истинная степень диссоциации для сильных электролитов составляет 100 % (a= 1). Однако из-за высокой концентрации ионов в растворах сильных электролитов проявляются силы межионного взаимодействия, поэтому определяемая опытным путем степень диссоциации не достигает максимального значения (100 %) и называется «кажущейся». Если величина «кажущейся» степени диссоциации неизвестна, то при расчетах степень диссоциации сильного электролита следует принять равной 1.
В растворах слабых электролитов процесс диссоциации протекает обратимо. Уравнение реакции диссоциации можно в общем виде представить как
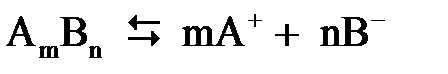 . .
| (2.8) |
Константа равновесия для процесса диссоциации слабого электролита называется константой диссоциации (КД). На основании закона действия масс выражение для константы равновесия (КС) или в данном случае константы диссоциации (КД) имеет вид
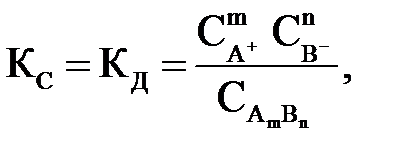
| (2.9) |
где 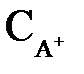 ,
, 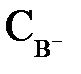 – равновесные концентрации ионов в растворе электролита, моль/л;
– равновесные концентрации ионов в растворе электролита, моль/л;  – концентрация недиссоциированных молекул, моль/л.
– концентрация недиссоциированных молекул, моль/л.
Константа диссоциации (КД) зависит от природы диссоциирующего вещества, растворителя, температуры и не зависит от концентрации раствора. Она характеризует способность электролита распадаться на ионы. Чем больше численное значение КД, тем в большей степени диссоциирует электролит.
Взаимосвязь между КД и aустанавливается законом разбавления Оствальда:
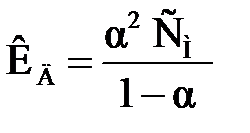 , ,
| (2.10) |
где СМ – молярная концентрация раствора электролита, моль/л.
Для слабых электролитов (a << 1) это выражение упрощается:
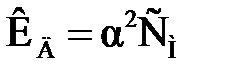 или или  . .
| (2.11) |
Закон разбавления Оствальда (уравнения (2.10) и (2.11)) выражает зависимость степени диссоциации слабого электролита от концентрации раствора: при уменьшении концентрации (разбавлении раствора) степень диссоциации увеличивается
б) молярная концентрация, или молярность (СМ или М), – число молей растворенного вещества в 1 л раствора:
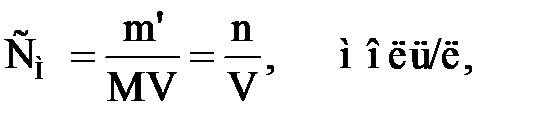
| (2.2) |
где 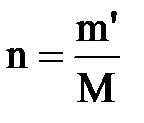 – количество вещества, моль; М – мольная масса растворенного вещества, г/моль; V – объем раствора, л.
– количество вещества, моль; М – мольная масса растворенного вещества, г/моль; V – объем раствора, л.
Например: 2 М раствор H2SO4 содержит в 1 л раствора 2 моля Н2SO4, или 2 × 98 = 196 г (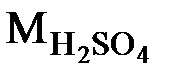 = 98 г/моль);
= 98 г/моль);
13.Диссоциация воды, α и Кд воды. Ионное произведение воды. Водородный и гидроксидный показатели (рН и рОН) и их взаимосвязь.(стр.20)
Ионное произведение воды. Водородный показатель
Вода является слабым электролитом, поэтому в любом водном растворе существует равновесие
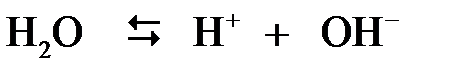 ,
,
которое количественно характеризуется константой диссоциации:
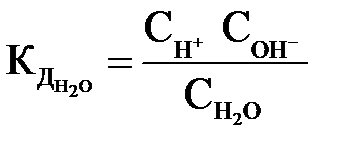 =1,86·10–16 при 298 К.
=1,86·10–16 при 298 К.
Численное значение 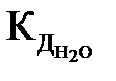 определено экспериментально по данным электропроводности воды. Равновесная концентрация недиссоциированных молекул воды
определено экспериментально по данным электропроводности воды. Равновесная концентрация недиссоциированных молекул воды 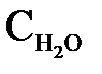 практически равна ее молярной концентрации, которая для воды объемом 1 л составляет
практически равна ее молярной концентрации, которая для воды объемом 1 л составляет  моль/л (1000/18) и является постоянной величиной. Произведение двух постоянных КД и дает новую постоянную, называемую константой воды
моль/л (1000/18) и является постоянной величиной. Произведение двух постоянных КД и дает новую постоянную, называемую константой воды 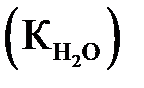 или ионным произведением воды:
или ионным произведением воды:
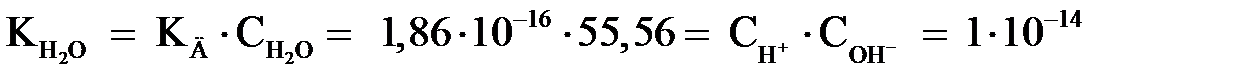 . .
| (2.13) |
Величина ионного произведения воды (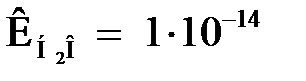 ) остается постоянной при неизменной температуре (298 К) не только в чистой воде, но и в любом водном растворе электролита (кислоты, основания, соли). Тогда
) остается постоянной при неизменной температуре (298 К) не только в чистой воде, но и в любом водном растворе электролита (кислоты, основания, соли). Тогда
 ; ; 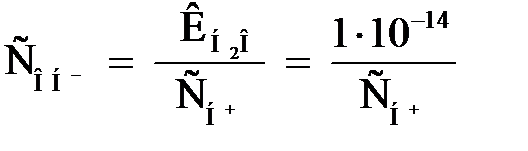 . .
| (2.14) |
В чистой воде 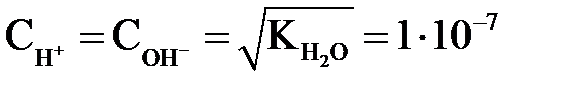 моль/л.
моль/л.
Для практической оценки характера среды в водных растворах используется водородный показатель рН.
Водородный показатель (рН) – отрицательный десятичный логарифм молярной концентрации ионов водорода в растворе
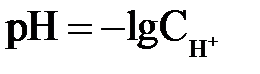 . .
| (2.15) |
Гидроксильный показатель (рОН) – отрицательный десятичный логарифм молярной концентрации гидроксид-ионов в растворе
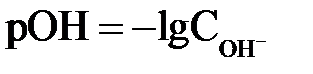 . .
| (2.16) | ||
| Тогда при 298 К | 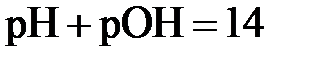 . .
| (2.17) | |
Таблица 2.1
Значение рН и рОН в различных средах при стандартных условиях
| Среда | 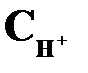 , моль/л , моль/л
| 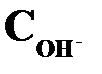 , моль/л , моль/л
| рН | рОН |
| Кислая | > 1 · 10–7 до 1 | < 1 · 10–7 | < 7 | > 7 |
| Нейтральная | 1 · 10–7 | 1 · 10–7 | ||
| Щелочная | < 1 · 10–7 | >1 · 10–7 | > 7 | < 7 |
14.1-й закон термодинамики. Внутренняя энергия (U) и энтальпия системы (Н) и их взаимосвязь. Физический смысл.
Термодинамика- наука, изучающая связи между превращениями в-ва и превращениями энергии. Термодинамика изучает возможность или невозможность самопроизвольного перехода системы из одного состояния в другое и энерг эффекты этих переходов. Знание хим.термодинамики позволяет: 1)предсказать условия устойчивости веществ и возможность их взаимодействия; 2)рассчитать тепловые балансы ф-х процессов; 3) раскрыть закономерности наблюдаемые при равновесии; 4) выбрать оптимальные режимы процесса Т, Р, С и др. хар.; 5) определить пути подавления или полного прекращения нежелательных реакций.
Функции состояния: т-д состояние систему описывается функциями состояния, которые определяются через параметры состояния P, T, V. Изменение хотя бы одного из них влечет изменение всех функций состояния. Функции состояния не зависят от пути и времени процесса, приводящие систему в данное состояние, а определяются только начальным и конечным состоянием системы. К функциям состояния относят: U внутренняя энергия; Н энтальпия;
Первый закон т/д. Внутренняя энергия, энтальпия: определение, физический смысл. Первый закон т-д: Теплота и работа – это два различных и единственно возможных способа обмена внутренней энергией между системой и средой при условиях, что переход вещества через границу системы отсутствует. ∆U=Q-A.
Первый закон т-д: Теплота, подведенная к системе, расходуется на увеличение внутренней энергии и на совершение системой механической работы. Q=∆U+A,A=p∆V+Amax, Q=∆U+p∆V+Amax
p∆V – работа расширения или работа против сил внешнего давления, необходимая для удержания системы в состоянии равновесия.
Amax – максимально полезная работа, которую может совершить система при протекании в ней самопроизвольных процессов.
Для химической реакции наиболее характерна работа расширения, т.е. Amax=0 Q=∆U+p∆V.
U внутренняя энергия: любая система, состоящая их большого числа частиц, находящихся в постоянном движении, характеризуется определенным запасом энергии. E=K+П+U (полная энергия системы). В т-д рассматриваются системы, находящиеся в покое (К=0) и пренебрегают действием силовых полей (П=0). Тогда Е=U.
| Внутренняя энергия системы – это ее полная энергия, состоящая из кинетической и потенциальной энергии всех частиц системы. Величина экстенсивная. Учет всех состояний при определении U невозможен, поэтому при т-д изучении системы достаточно знать изменение ее внутренней энергии при переходе из состояния U1 в U2. ∆U=U2-U1. Теплота. Работа: Изменение внутренней энергии может происходить за счет обмена энергией между системами или системой и средой в двух формах: теплоты и работы. Теплота – мера переданной энергии за счет хаотического столкновения молекул соприкасающихся тел. Теплота стимулирует хаотическое движение. Работа – мера переданной энергии путем упорядоченного перемещения вещества от одной системы к другой под действием каких-либо сил. Работа стимулирует организованное движение. Теплота и работа относятся не к состоянию системы, а к какому-то процессу, т.е. являются функциями процесса. Энтальпия(теплосодержание системы)- сумма внутр энергии системы и работы,которую она производит против силы внешнего давления(есть полная энергия расширенной системы):это ф-ция состояния: H=U+PV [Дж] Физический смысл энтальпии:полная энергия расширенной системы в изобарно-изотермических условиях: ∆H=∆U+P∆U |
15.Стандартные энтальпии образования сложных соединений из простых (ΔН0298). Закон Гесса и следствие из него.
Стандартная энтальпия образования соединения ∆Hоf,298 –энтальпия р-ции образования 1 моля этого соединения в ст.условиях из простых в-в, находящихся в станд состоянии.Размерность кДж/моль. Стандартные условия Р=101.3кПа Т=298
| Стандартные состояния вещества – физическое состояние, в котором чистое вещество наиболее стабильно при стандартных условиях. Энтальпия образования простых веществ в стандартном состоянии принята равной 0. |
Расчет тепловых эффектов химических процессов. Уравнение Кирхгофа. Для расчета тепловых эффектов используются основные законы т-х.:
1)Закон Лавуазье-Лапласа: теплота которая выделяется при образования хим соединения равна теплоте поглощаемой при его разложении на исходные в-ва(тепловой эффект прямой р-ции равен и противоположен по знаку тепловому эффекту обратной р-ции. 2) Закон Гесса – тепловой эффект реакции не зависит от пути процесса, определяется только начальным и конечным состояниями веществ при условии что система совершает только механическую работу. Следствие: стандартная энтальпия реакции равна разности стандартных энтальпий образования продуктов реакции и исходных веществ с учетом стехиометрических коэффициентов в уравнении реакции.
Характеристика индивидуального вещества – ∆Hof,298
Характеристика процесса – ∆Ho298
Численное значение теплового эффекта любой р-ции зависит от:природы реаг в-в и их агрегатного состояния.от числа молей. От температуры.
Для расчета теплового эффекта при любой исходной температуре используется уравнение Кирхгофа:
∆HоT =∆Ho298+∆Cop,298*(T-298)
Cop - Молярная изобарная теплоемкость кол-ва теплоты, необходимой для нагревания 1 моля вещества на 1К при постоянном давлении. Условно принято, что теплоемкость не зависит от температуры, поэтому можно рассмотреть изменение теплоемкости в ходе процесса.
Изменении энтальпии характеризует вещество и процесс, но тепловой эффект нельзя использовать в качестве критерия осуществимости процесса.
16.Расчет тепловых эффектов х. р. при стандартных условиях и при заданной температуре (ΔН0298 (х. р.) и ΔН0Т (х. р.)).
17.2-ой закон термодинамики. Энтропия системы (S) и её физический смысл. Стандартные энтропии веществ (S0298).
Второй закон термодинамики определяет направленность превращения энергии при переходе системы из одного состояния в другое и имеет несколько эквивалентных формулировок в зависимости от группы явлений, к которым применяется.
1)Теплота не может самопроизвольно переходить от менее нагретого тела к более нагретому.(Клаузиус)
2)Невозможно всю теплоту превратить в полную работу, часть ее непременно теряется. (Кельвин-Планк) Q=T∆S (Q – рассеянная энергия, та часть теплоты, которая не может быть превращена в работу и определяет меру беспорядка в системе, ∆S – изменение энтропии). Термодинамический смысл энтропии – мера связанной энергии, отнесенная к 1К, т.е. мера перехода энергии в такую форму, из которой она не может самостоятельно переходить в другие формы.
3)Естественные процессы развиваются необратимо в направлении увеличения беспорядка (Больцман). S=К ln W – уравнение Больцмана. К- постоянная Больцмана, W- термодинамическая вероятность состояния системы – число микросостояний системы, которым можно реализовать данное макросостояние.
Статистический смысл энтропии – мера неупорядоченности системы, количественная мера хаотического движения и взаимного расположения частиц системы.
В изолированных системах энтропия самопроизвольно протекающего процесса возрастает, в случае неизолрованных систем энтропия в ходе процесса может как увеличиваться так и уменьшаться:
Увеличение-расширение газов,фазовые превражение (т-ж-г),растворение кристалических в-в
Уменьшение-сжатие газов,конденсация и кристализация в-в (г-ж-т)
В отличие от других т-д функций для любого вещества можно определить абсолютное значение энтропии, т.к каждое вещество в определенном агрегатном состоянии имеет свой собственный «беспорядок», обусловленный его природой.
Стандартная энтропия вещества (So298) – это энтропия 1 моля вещества в его стандартном состоянии при стандартных условиях. Значения в таблице.[Дж/мольК]
18.Он
19.Расчет изменения энтропии химических реакций при ст. у. (ΔS0298 (х. р.)) и при заданной температуре (ΔS0 Т (х. р.)).
20.Уравнение, объединяющее 1-й и 2-й законы термодинамики. Свободная энергия Гиббса (G) и её физический смысл.
| Уравнение, объединяющее первый и второй законы т/д. Свободная энергия Гиббса или изобарно-изотермический потенциал (G) и его изменение (ΔG) в физико-химических процессах при стандартных условиях и заданной температуре Т. Вывод основного уравнения т-д Первый закон Q=∆U+p∆V+Amax Второй закон Q=T∆S T∆S=∆U+p∆V+Amax=∆H+Amax Amax= T∆S-∆H= (TS2-TS1)-(H2-H1)= (H1-TS1)-(H2-TS2) Функция (H-TS) – энергия Гиббса Amax= G1-G2= -(G2-G1)= - ∆G Основное уравнение т-д ∆G=∆H-T∆S Физический смысл ∆G – это та часть энергетического эффекта реакции, которую можно превратить в работу в обратном процессе при P,T=const Свободная энергия Гиббса: Из основного уравнения т-д следует 2-ой подход в определении физического смысла энергии Гиббса. Разность между полной энергией расширенной системы и ее связанной энергией есть свободная энергия системы ∆G. |
Расчет ∆G в ходе реакции: стандартные условия
21.Свободная и связанная энергии системы. Роль энтальпийного и энтропийного факторов в определении направленности протекания самопроизвольных процессов.
22.Термодинамические условия протекания обратимых х. р. и наступления равновесия. Расчет температуры равновесия.
| Термодинамическое условие равновесия Пределом убыли свободной энергии Гиббса при самопроизвольном протекании процесса является ее минимальное значение, отвечающее состоянию равновесия. Такое состояние системы наиболее устойчиво, всякое отклонение от него требует затрат энергии. ∆G=0, ∆H-T∆S=0 В состоянии термодинамического равновесия оба процесса в системе равновероятны. Пренебрегая зависимостью ∆H и ∆S от температуры можно приближенно определить температуру наступления равновесия: Tравн=∆Ho298/∆So298 |
Связь между ∆G и Kp
| Взаимосвязь между константой равновесия и изобарным потенциалом выражается уравнением изотермы хим р-ции. ____________________________________ |
Зная ∆G можно по уравнению изотермы рассчитать Kp, а значит установить предел протекания процесса должном направлении.
23.Электрохимические процессы и системы. Понятие электродов, природа возникновения электродных потенциалов (φ, В).(стр.31)
Электрохимические процессы можно разделить на две основные группы:
1) процессы превращения химической энергии самопроизвольно идущих окислительно-восстановительных реакций в электрическую (в гальванических элементах);
2) процессы превращения электрической энергии в химическую (электролиз).
Простейшая электрохимическая система состоит из двух электродов и ионного проводника между ними. Электроды замыкаются металлическим проводником. Ионным проводником (проводником 2-го рода) служат растворы, расплавы электролитов или твердые электролиты.
Электрод – система, состоящая из двух контактирующих фаз: материала с электронной проводимостью и ионного проводника.
Электроды бывают:
1) инертные (нерастворимые), не способные окисляться в ходе реакции, например графитовые или платиновые;
2) активные (растворимые), способные окисляться в ходе реакции. В общем случае это любой металл, кроме благородного. В зависимости от условий эксплуатации некоторые активные металлы могут вести себя как инертные из-за явления пассивации.
Все металлы характеризуются свойством в большей или в меньшей степени растворяться в воде. Под действием полярных молекул (диполей) воды ионы поверхностного слоя металла отрываются и в гидратированном состоянии переходят в жидкость. При этом пластина металла заряжается отрицательно (из-за появления в ней избыточных электронов), а слой электролита у ее поверхности – положительно (гидратированные катионы металла). В результате электростатического притяжения противоположных зарядов на границе металл – раствор возникает двойной электрический слой иразность потенциалов в нем. С увеличением концентрации катионов у поверхности металла становится вероятным обратный процесс – восстановление ионов металла. Когда скорости указанных процессов сравняются, в системе устанавливается равновесие, которое можно выразить уравнением
Me + 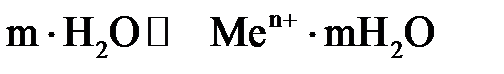 + + 
| (3.1) |
Состояние равновесия зависит от активности металла, концентрации его ионов в растворе и численно характеризуется константой равновесия, которая для данной гетерогенной системы при P = const равна
 . .
| (3.2) |
Для активных металлов(Zn, Fe, Cr и др.) равновесие(3.1) смещено вправо (КР>1). Например, Zn D 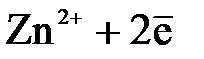 (
(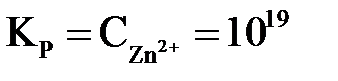 ). При погружении их в водный раствор собственной соли для достижения равновесной концентрации ионы металла будут переходить в раствор и поверхность металла зарядится отрицательно, а раствор электролита – положительно.
). При погружении их в водный раствор собственной соли для достижения равновесной концентрации ионы металла будут переходить в раствор и поверхность металла зарядится отрицательно, а раствор электролита – положительно.
Для малоактивных металлов (Cu, Ag, Hg и др.) равновесная концентрация ионов металла в растворе очень мала и равновесие (3.1) смещается влево (КР<1). Например, Cu D 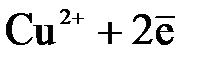
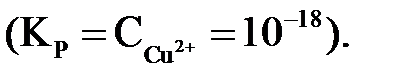 При погружении их в водный раствор собственной соли для достижения равновесной концентрации ионы металла из раствора будут переходить на поверхность металла и она зарядится положительно, а раствор электролита – отрицательно.
При погружении их в водный раствор собственной соли для достижения равновесной концентрации ионы металла из раствора будут переходить на поверхность металла и она зарядится положительно, а раствор электролита – отрицательно.
Потенциал, возникающий на металлическом электроде, находящемся в равновесии с собственными ионами в растворе электролита, называется электродным потенциалом (φ, В).
24.Металлические и газовые электроды, их схематическая запись. Уравнения электродных реакций.(стр.32)
Электроды, обратимые относительно своих ионов в растворе электролита, называются электродами 1-го рода. К ним относятся металлические и газовые электроды: водородный, кислородный.
| Схема | 
|
| Уравнения электродных реакций | 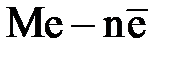 D D 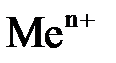 , , 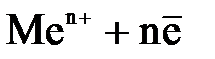 D D 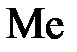
|
|
|
|
|
|
Дата добавления: 2015-04-24; Просмотров: 767; Нарушение авторских прав?; Мы поможем в написании вашей работы!