КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Комплексонометрия
|
|
|
|
Метод Фаянса. Прямая аргентометрия.
Титрант – AgNO3, индикаторы – эозинат натрия (Br–,I–), бромтимоловый синий (Cl–), среда – CH3COOH (30%).
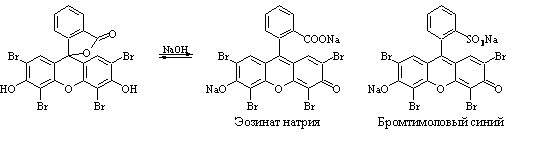
NaI + AgNO3 → AgI↓ + NaNO3.
Йодид серебра адсорбирует на себе одноименные ионы; появляется ярко-розовая окраска:
{ m (AgI)∙ n I–(n – x)K+} x–
В точке эквивалентности коллоидная частица становится электронейтральной, в КТТ начинает адсорбировать Ag+; идет перезарядка мицеллы, осадок коагулирует, раствор просветляется:
{ m (AgI)∙ n I–(n – x)K+} x– ∙ x Ag+,
{ m (AgI)∙ n I–(n – x)K+} x– ∙ x Ag+ + Ind2– → (x /2)Ag2Ind + { m (AgI)∙ n I–(n – x)K+} x–.
f экв(NaI)=1, 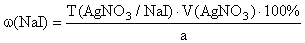
Комплексонометрия, хелатометрия, трилонометрия, метод титриметрического анализа, основанный на образовании хорошо растворимых в воде и слабо диссоциированных комплексных соединений при реакции большинства катионов с аминополикарбоновыми кислотами (комплексонами). Конечную точку титрования устанавливают с помощью комплексонометрических и др. индикаторов (см. Индикаторы химические). В некоторых случаях, особенно при титровании мутных или сильно окрашенных растворов, используют безиндикаторные (инструментальные) методы — потенциометрию, кулонометрию, кондуктометрию (см. Электрохимические методы анализа), фотометрический анализ и др. К. применяют для определения содержания металлов в минеральном сырье, продуктах металлургического производства, фармацевтических препаратах, определения жёсткости воды и др.
28. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
основаны на зависимости физ. св-в в-ва от его природы, причем ана-лит. сигнал представляет собой величину физ. св-ва, функционально связанную с концентрацией или массой определяемого компонента. Ф.-х. м. а. могут включать хим. превращения определяемого соед., растворение образца,концентрирование анализируемого компонента, маскирование мешающих в-в и др. В отличие от "классич." химических методов анализа, где аналит. сигналом служит масса в-ва или его объем, в Ф.-х. м. а. в качестве аналит. сигнала используют интенсивность излучения, силу тока, электропроводность, разность потенциалов и др.
Важное практич. значение имеют методы, основанные на исследовании испускания и поглощения электромагн. излучения в разл. областях спектра. К ним относится спектроскопия (напр., люминесцентный анализ, спектральный анализ}, нефелометрия и турбидиметрия и др. К важным Ф.-х. м. а. принадлежат электрохим. методы, использующие измерение электрич. св-в в-ва (вольтамперометрия, кондуктометрия, кулонометрия,потенциометрия и т. д.), а также хроматогра-фия (напр., газовая хроматография, жидкостная хроматог-рафия, ионообменная хроматография, тонкослойная хроматография). Успешно развиваются методы, основанные на измерении скоростей хим. р-ций (кинетические методы анализа), тепловых эффектов р-ций (термометрич. титрование, см. Калориметрия), а также на разделении ионов в магн. поле (масс-спектрометрия).
При выполнении Ф.-х. м. а. используют специальную, иногда довольно сложную, измерит. аппаратуру, в связи с чем эти методы часто наз. инструментальными. Многие совр. приборы оснащены встроенными ЭВМ, к-рые позволяют находить оптим. условия анализа (напр., спектральную область получения наиб. точных результатов при анализе смеси окрашенных в-в), выполняют расчеты и т. д.
Почти во всех Ф.-х. м. а. применяют два основных приема: методы прямых измерений и титрования. В прямых методах используют зависимость аналит. сигнала от природы анализируемого в-ва и его концентрации. Зависимость сигнала от природы в-ва - основа качеств. анализа (потенциал полуволны в полярографии и т. д.). В нек-рых методах связь аналит. сигнала с природой в-ва установлена строго теоретически. Напр., спектр атома водорода м. б. рассчитан по теоретически выведенным ф-лам. В количеств. анализе используют зависимость интенсивности сигнала от концентрации в-ва. Чаще всего она имеет вид I = a + bс (ур-ние связи), где I- интенсивность сигнала (сила диффузионного тока в полярографии, оптич. плотность в спектрофотометрии и т. д.), с - концентрация, аи b - постоянные, причем во мн. случаях а = 0 (спектрофотометрия, полярография и др.). В ряде Ф.-х. м. а. ур-ние связи установлено теоретически, напр. закон Бугера-Ламберта-Бера (фотометрический анализ), ур-ние Ильковича (вольтамперометрия).
Численные значения констант в ур-нии связи определяют экспериментально с помощью стандартных образцов, стандартных р-ров и т. д. Только в кулонометрии, основанной на законе Фарадея, не требуется определение констант.
Наиб. распространение в практике получили след. методы определения констант ур-ния связи или, что то же самое, методы количеств, анализа с помощью физ.-хим. измерений:
1) Метод градуировочного графика. Измеряют интенсивность аналит. сигнала у неск. стандартных образцов или стандартных р-ров и строят градуировочный график в координатах I= f(с) или I= f(lgc), где с - концентрация компонента в стандартном р-ре или стандартном образце. В тех же условиях измеряют интенсивность сигнала у анализируемой пробы и по градуировочному графику находят концентрацию.
2) Метод молярного св-ва применяют в тех случаях, когда ур-ние связи I = bc соблюдается достаточно строго. Измеряют аналит. сигнал у неск. стандартных образцов или р-ров и рассчитывают b= I ст /с ст; если с ст измеряется в моль/л, то b - молярное св-во. В тех же условиях измеряют интенсивность сигнала у анализируемой пробы x и по соотношению <cx</c= x /b> или <cx = c></c стx /I> СТ рассчитывают концентрацию.
3) Метод добавок. Измеряют интенсивность аналит. сигнала пробы x,> а затем интенсивность сигнала пробы с известной добавкой стандартного р-ра x+ стt. Концентрацию в-ва в пробе рассчитывают по соотношению с x= с стx/(Ix+ ст< - Ix)>.
Методы титрования. Измеряют интенсивность аналит. сигнала I в зависимости от объема Vдобавленного титранта. По кривой титрования I=f (V)находят точку эквивалентности и рассчитывают результат по обычным ф-лам титриметрич. анализа (см. Титриметрия).
Ф.-х. м. а. часто используют при определении низких содержаний (порядка 10-3% и менее), где-классич. хим. методы анализа обычно неприменимы. В области средних и высоких концентраций хим. и Ф.-х. м. а. успешно конкурируют между собой, взаимно дополняя друг друга. Ф.-х. м. а. развиваются в направлении поиска новых хим.-аналит. св-в в-ва, увеличения точности анализа, конструирования новых прецизионных аналит. приборов, совершенствования существующих методик и автоматизации анализа. Интенсивно развивается в последнее время проточно-ижкекционный анализ - один из наиб. универсальных вариантов автоматизир. анализа, основанный на дискретном введении микрообъемов анализируемого р-ра в поток жидкого носителя с реагентом и последующего детектирования смеси тем или иным физ.-хим. методом.
Деление аналит. методов на физ., хим. и физ.-хим. весьма условно. Часто к Ф.-х. м. а. относят, напр., ядерно-физ. методы. В последнее время наметилась тенденция делить методы анализа на хим., физ. и биол.- вовсе без физ.-химических.
29. Оптические методы исследования
основаны на использовании законов оптики, касающихся природы, распространения и взаимодействия с веществом электромагнитного излучения оптического диапазона (видимый свет, ультрафиолетовое и инфракрасное излучение).
Законы геометрической оптики, характеризующие прямолинейное распространение света в однородных средах, его отражение и преломление в гетерогенных средах, лежат в основе расчета, конструирования и эксплуатации таких широко используемых в медицине приборов, как микроскопы, рефрактометры, медицинские осветители, аппараты для светолечения, эндоскопы, лазерные установки и др.
Для качественного и количественного определения химических элементов в биологических жидкостях и тканях, в лекарственных препаратах и других объектах служит спектрально-эмиссионный анализ. Он заключается в изучении спектра света, который испускают атомы и молекулы, возбужденные различными способами, например нагреванием до высоких температур. Разновидностью эмиссионного анализа является метод пламенной фотометрии, позволяющий определять содержание в биологических образцах ионов калия, натрия, лития и др.
Действие большой группы оптических приборов основано на оптических законах взаимодействия света с веществом. Для измерения рефракции или показателя преломления света исследуемых образцов используют рефрактометры. Их применяют при определении чистоты дистиллированной воды, содержания сахарозы в водных растворах, общего белка в сыворотке крови и пр.
Для измерения поглощения света веществом с целью анализа состава и структуры образца широко применяют фотометрические и спектрофотометрические методы (колориметрию, фотометрию, спектрофотометрию). Приборы, служащие для этой цели, получили название колориметров, фотометров, спектрофотометров. Спектрофотометры позволяют изучать характерные спектры поглощения различных веществ и устанавливать их химическое строение и количественное содержание в растворах, например ферментов, гормонов, витаминов, нуклеиновых кислот, углеводов, спиртов, липидов и др.
Величина рассеяния света исследуемыми объектами, например коллоидными растворами, определяется методом нефелометрии и турбидиметрии. О содержании вещества судят по интенсивности светового потока, который рассеивается взвешенными частицами определяемого вещества (нефелометрия), или по поглощению светового потока этими частицами (турбидиметрия).
Для измерения концентрации и изучения свойств оптически активных молекул применяют поляриметры и спектрополяриметры. В медицине они находят наибольшее практическое применение при определении концентрации сахара в моче (сахарометрия).
Поглощение света веществом нередко сопровождается возникновением вторичного излучения с меньшей длиной волны (см. Люминесценция). Приборы, служащие для изучения люминесценции, получили название флюориметров, спектрофлюориметров, микроцитоспектрофлюориметров. Они используются для структурного анализа биологически активных молекул, определения количественного содержания в образце витаминов, ферментов, промежуточных продуктов обмена, стероидов.
Совокупность оптических методов, используемых для изучения процесса восприятия света глазом человека, традиционно выделяется в самостоятельный раздел оптики — физиологическую оптику. Они служат для исследования остроты зрения, абсолютной чувствительности, критической частоты слияния световых мельканий и др. В этих методах используют такие оптические приборы, как офтальмоскопы, офтальмометры, глазные рефрактометры, адаптометры и т.д.
30. Колориметрия (от лат. color — цвет и греч. μετρώ — измеряю) — физический метод химического анализа, основанный на определении концентрации вещества по интенсивности окраски растворов (более точно — по поглощению света растворами).
Колориметрия — это метод количественного определения содержания веществ в растворах, либо визуально, либо с помощью приборов, таких какколориметры.
Колориметрия может быть использована для количественного определения всех тех веществ, которые дают окрашенные растворы, или могут быть, с помощью химической реакции, дать окрашенное растворимое соединение. Колориметрические методы основываются на сравнении интенсивности окраскиисследуемого раствора, изучаемого в пропущенном свете, с окраской эталонного раствора, содержащего строго определенное количество этого же окрашенного вещества, или же с дистиллированной водой.
Любопытна история возникновения колориметрии и фотометрии. Ю. А. Золотов упоминает, что Роберт Бойль (так же, как и некоторые ученые до него) использовал экстракт дубильных орешков, чтобы различить железо и медь в растворе. Однако, по-видимому, именно Бойль впервые заметил, что чем больше железа содержится в растворе, тем более интенсивна окраска последнего. Это был первый шаг к колориметрии. А первым инструментом колориметрии стали колориметры типа колориметра Дюбоска (1870)[1], которые использовались вплоть до недавнего времени[2].
Более совершенные приборы — спектрофотометры — отличаются возможностью исследования оптической плотности в широком диапазоне длин волнвидимого спектра, а также в ИК и УФ-диапазонах, с меньшей дискретностью длины волны (с использованием монохроматора).
Фотоколориметры и спектрофотометры измеряют величину пропускания света при определенной длине волны света. Контроль (обычно дистиллированная вода или исходный материал без добавления реагентов) используется для калибровки устройства.
Колориметрия широко применяется в аналитической химии, в том числе для гидрохимического анализа, в частности — для количественного анализа содержания биогенных веществ в природных водах[3], для измерения pH[4], в медицине, а также в промышленности при контроле качества продукции.
32. Потенциометрия
(от лат. potentia - сила, мощность и греч. metreo - измеряю * a. potentiometry; н. Potentiometrie; ф. Potentiometrie; и. potenciometria) - электрохим.метод исследования и анализа веществ, основанный на измерении электродвижущих сил (эдс) обратимых гальванич. элементов. П. используется для изучения кинетики и определения констант устойчивости комплексных соединений, констант диссоциации слабых кислот и оснований, а также произведения растворимости малорастворимых электролитов. П. включает редоксметрию и ионометрию. Гальванич. элемент, применяемый в П., обычно состоит из 2 электродов, к-рые помещают или в один и тот же раствор (элемент без переноса), или в 2 различных по составу раствора, имеющих между собой жидкостный контакт (элемент с переносом). Элементы без переноса используют для определения изменений термодинамич. потенциалов, энтропий, энтальпий и др. величин при разл. процессах.
Электрод, по потенциалу к-рого судят о концентрации определяемых ионов в растворе, наз. индикаторным. Величину потенциала индикаторного электрода определяют, сравнивая её с величиной потенциала др. электрода, к-рый принято называть электродом сравнения. В качестве электрода сравнения может быть применён только такой электрод, величина потенциала к-рого остаётся неизменной при изменении концентрации определяемых ионов.
Различают прямую П. и потенциометрич. титрование (ПТ). Первая используется для непосредственного определения концентрации веществ по значению потенциала индикаторного электрода (рН-метрия, ионометрия). При ПТ в исследуемый раствор, помещённый в потенциометрич. ячейку, опускают индикаторный электрод, возникновение потенциала на к-ром обусловливается или непосредственно определяемым веществом (если оно электроактивно), или косвенно (если оно неэлектроактивно) в результате его хим. взаимодействия с др. потенциалопределяющим компонентом. Конечную точку титрования (КТТ) определяют по скачку потенциала, вызванного заменой одной электрохим. реакции другой до и после КТТ с соответствующим изменением величины потенциала. Этот вариант метода называют ПТ без тока. КТТ удобно определять графически по кривой титрования (рис.).
При работе с необратимыми редокс-системами или компонентами обратимых редокс-систем (в отсутствие сопряжённых форм) потенциал индикаторного электрода устанавливается медленно, он неустойчив. В таких случаях применяют ПТ под током, к-рое заключается в том, что индикаторный электрод поляризуется, т.е. через него с помощью внеш. источника тока пропускают ток малой величины (неск. микроампер). В этом методе можно использовать 2 идентичных индикаторных электрода, погруженных в титруемый раствор, один из к-рых поляризуют анодно, а другой - катодно. При этом отпадает необходимость в электроде сравнения, а измеренная эдс соответствует разности потенциалов между 2 электродами. Ход титрования описывается дифференциальной кривой. В ПТ применяют реакции нейтрализации, окисления- восстановления, комплексообразования, осаждения. При использовании техники ультрамикроанализа с помощью ПТ можно определять - n * 10-10 веществ, напр., до 0,4 нг Сu2+ с относительным стандартным отклонением 4-9%. Достоинства ПТ: низкие границы определяемых концентраций, объективность и точность установления КТТ, селективность, возможность титрования в окрашенных и мутных средах, последовательное титрование неск. компонентов, простота автоматизации. П. широко применяется для анализа пром. и природных материалов, напр. руд, минералов, т.п., а также объектов окружающей среды. П. используется также в автоматизир. системах аналитич. контроля технол. потоков на обогатит. ф-ках и гидрометаллургич. з-дах.
33. Полярография — один из важнейших электрохимических методов анализа веществ, исследования кинетики химических процессов. Принцип метода[править]
Протекание электрического тока в водном растворе связано с движением ионов, образованных в результате электролитической диссоциации. Протекание тока через ртуть, другие металлические и углеродные материалы – с движением электронов. Поэтому на границе электрод/раствор должен существовать какой-то процесс, обеспечивающий переход потока ионов в поток электронов, иначе ток не пойдет. Такой процесс представляет собой электрохимическую реакцию. Количество прореагировавшего вещества определяется законом Фарадея, то есть пропорционально прошедшему через электрод заряду:
М = Мэкв * Q/zF, (1)
Где М – масса прореагировавшего вещества, Мэкв – эквивалентная масса прореагировавшего вещества, Q - прошедший через электрод заряд, z- количество электронов, участвующих в превращении одной молекулы или одного иона, F- число Фарадея, задающее коэффициент пропорциональности. Число Фарадея равно 96485 кулон/моль и представляет собою число Авогадро, умноженное на заряд электрона. Если отнести уравнение (1) к единице времени, масса превратится в массовую скорость реакции (поток вещества) J, а заряд – в ток i, которые обычно относят к единице поверхности электрода (плотность тока): J = Мэкв * i/zF,
Метод основан на анализе кривых зависимостей силы тока от приложенного к электрохимической ячейке напряжения — так называемых полярограмм. В зависимости от формы и скорости изменения поляризующего напряжения различают постояннотоковую (классическую), переменнотоковую, высокочастотную, импульсную, осциллографическую полярографию, варианты метода имеют различные чувствительность (минимально определяемая концентрация вещества) и разрешающую способность (допустимое отношение концентраций определяемого компонента и сопутствующих).
В ячейке для полярографии присутствуют поляризуемый и неполяризуемый электроды, площадь первого должна быть значительно меньше площади второго — в таком случае идущая на нём электродная реакция не вызывает заметных химических изменений в растворе или изменения разности потенциалов. В качестве поляризуемого электрода могут быть использованы ртутно-капающий электрод, стационарный ртутный электрод, твёрдые электроды из графита, благородных металлов и пр.
34. АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
метод количеств. анализа, основанный на волътамперометрии с линейной разверткой потенциала. Конечную точку титрования устанавливают по зависимости диффузионного тока d при постоянном потенциале Е с индикаторного электрода от объема Vприбавленного титранта. Электрохимически активным в-вом, обусловливающим измеряемый диффузионный ток, м. б. определяемый компонент, титрант, продукт их взаимод. или в-во ("индикатор"), дополнительно введенное в электролитич. ячейку. Выбор значения Е с производят по вольтамперограммам определяемого в-ва (см. рис.) и титранта. Титрантом служит р-р реагента - осадителя, окислителя, восстановителя или комплексообразующего в-ва, концентрация к-рого на неск. порядков превышает концентрацию определяемого в-ва, с к-рым он взаимодействует. Титрант прибавляют из микробюретки небольшими порциями, благодаря чему разбавлением исследуемого р-ра можно пренебречь.
Форма кривых титрования d=f(V>)зависит от того, какое в-во электроактивно до и после точки эквивалентности, но всегда эти кривые имеют по крайней мере две ветви, к-рые пересекаются в конечной точке (к. т.) титрования.
АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
метод количеств. анализа, основанный на волътамперометрии с линейной разверткой потенциала. Конечную точку титрования устанавливают по зависимости диффузионного тока d при постоянном потенциале Е с индикаторного электрода от объема Vприбавленного титранта. Электрохимически активным в-вом, обусловливающим измеряемый диффузионный ток, м. б. определяемый компонент, титрант, продукт их взаимод. или в-во ("индикатор"), дополнительно введенное в электролитич. ячейку. Выбор значения Е с производят по вольтамперограммам определяемого в-ва (см. рис.) и титранта. Титрантом служит р-р реагента - осадителя, окислителя, восстановителя или комплексообразующего в-ва, концентрация к-рого на неск. порядков превышает концентрацию определяемого в-ва, с к-рым он взаимодействует. Титрант прибавляют из микробюретки небольшими порциями, благодаря чему разбавлением исследуемого р-ра можно пренебречь.
Форма кривых титрования d=f(V>)зависит от того, какое в-во электроактивно до и после точки эквивалентности, но всегда эти кривые имеют по крайней мере две ветви, к-рые пересекаются в конечной точке (к. т.) титрования.
35. Кулонометри́я — один из электрохимических методов анализа, применяется в аналитической химии, объединяет методы анализа, основанные на измерении количества вещества, выделяющегося на электроде в процессе электрохимической реакции в соответствии с законами Фарадея.
При кулонометрии потенциал рабочего электрода отличается от равновесного значения. Различают потенциостатическую и гальваностатическую кулонометрию, причём последняя включает прямой и инверсионный методы, электроанализ и кулонометрическое титрование.
36. Хроматогра́фия (от др.-греч. χρῶμα — цвет) — динамический сорбционный метод разделения и анализа смесей веществ, а также изучения физико-химических свойств веществ. Основан на распределении веществ между двумя фазами — неподвижной (твердая фаза или жидкость, связанная на инертном носителе) и подвижной (газовая или жидкая фаза, элюент). Название метода связано с первыми экспериментами по хроматографии, в ходе которых разработчик метода Михаил Цвет разделял ярко окрашенные растительные пигменты. Метод хроматографии был впервые применён русским учёным-ботаником Михаилом Семеновичем Цветом в 1900 году. Он использовал колонку, заполненную карбонатом кальция, для разделения пигментов растительного происхождения. Первое сообщение о разработке метода хроматографии было сделано Цветом 30 декабря 1901 года на XI Съезде естествоиспытателей и врачей в С.-Петербурге. Первая печатная работа по хроматографии была опубликована в 1903 году, в журнале Труды Варшавского общества естествоиспытателей. Впервые термин хроматография появился в двух печатных работах Цвета в 1906 году, опубликованных в немецком журнале Berichte der Deutschen Botanischen Gesellschaft. В 1907 году Цвет демонстрирует свой метод Немецкому Ботаническому обществу.
В 1910—1930 годы метод был незаслуженно забыт и практически не развивался.
В 1931 году Р. Кун, А. Винтерштейн и Е. Ледерер при помощи хроматографии выделили из сырого каротина α и β фракции в кристаллическом виде, чем продемонстрировали препаративную ценность метода.
В 1941 году А. Дж. П. Мартин и Р. Л. М. Синг разработали новую разновидность хроматографии, в основу которой легло различие в коэффициентах распределения разделяемых веществ между двумя несмешивающимися жидкостями. Метод получил название «распределительная хроматография».
В 1947 году Т. Б. Гапон, Е. Н. Гапон и Ф. М. Шемякин разработали метод «ионообменной хроматографии».
В 1952 году Дж. Мартину и Р. Сингу была присуждена Нобелевская премия в области химии за создание метода распределительной хроматографии.
С середины XX века и до наших дней хроматография интенсивно развивалась и стала одним из наиболее широко применяемых аналитических методов.
37. КОЛОРИМЕТРИЯ
(цветовые измерения), наука о методах измерения и количеств. выражении цвета. В результате цветовых измерений (ЦИ) определяются три числа, т. н. цветовые координаты (ЦК), полностью определяющие цвет при нек-рых строго стандартизованных условиях его рассматривания.
Цветовые координатные системы и цветность. Основой матем. описания цвета в К. явл. экспериментально установленный факт, что любой цвет при соблюдении упомянутых условий можно представить в виде смеси (суммы) определённых кол-в трёх линейно независимых цветов, т. е. таких цветов, каждый из к-рых не может быть представлен в виде суммы к.-л. кол-в двух других цветов. Групп (систем) линейно независимых цветов существует бесконечно много, но в К. используются лишь нек-рые из них. Три выбранных линейно независимых цвета наз. основными цветами (ОЦ); они определяют цветовую координатную систему (ЦКС). Тогда три числа, описывающие данный цвет, явл. кол-вами ОЦ в смеси, цвет к-рой зрительно неотличим от данного цвета; эти три числа и есть ЦК данного цвета.
Эксперим. результаты, к-рые кладут в основу разработки колорнметрич. ЦКС, получают при усреднении данных наблюдений (в строго определённых условиях) большим числом наблюдателей; поэтому они не отражают точно св-в цветового зрения к.-л. конкретного наблюдателя, а относятся к т. н. среднему стандартному колориметрич. наблюдателю. Будучи отнесены к стандартному наблюдателю в определённых неизменных условиях, стандартные результаты смешения цветов и построенные на их основе колориметрич. ЦКС описывают фактически лишь физ. аспект цвета, не учитывая изменения цветовосприятия глаза при изменении условий наблюдения, интенсивности цвета и по др. причинам (см. ЦВЕТ).
Когда ЦК к.-л. цвета откладывают по трём взаимно перпендикулярным координатным осям, этот цвет геометрически представляется точкой в трёхмерном, т. н. цветовом, пространстве (ЦП) или же вектором, начало к-рого совпадает с началом координат, а конец — с упомянутой точкой цвета. Точечная и векторная геом. трактовки цвета равноценны и обе используются в К. Точки, представляющие все реальные цвета, заполняют нек-рую область ЦП. Но математически все точки пр-ва равноправны, поэтому можно условно считать, что и точки вне области реальных цветов представляют нек-рые цвета. Такое расширение толкования цвета как матем. объекта приводит к понятию нереальных цветов, к-рые невозможно наблюдать или как-либо реализовать практически. Тем не менее с этими цветами можно производить матем. операции так же, как и с реальными цветами, что оказывается чрезвычайно удобным. За единичные кол-ва ОЦ в ЦКС принимают такие их кол-ва, к-рые дают в смеси нек-рый исходный (опорный) цвет (чаще всего белый).
Своего рода «качество» цвета, наз. его цветностью, геометрически удобно характеризовать в двумерном пр-ве — на «единичной» плоскости ЦП, проходящей через три единичные точки координатных осей (осей ОЦ). Линии пересечения единичной плоскости с координатными плоскостями образуют на ней т. н. цветовой треугольник, в вершинах к-рого находятся единичные значения ОЦ. Если такой треугольник— равносторонний, его часто наз. треугольником Максвелла. Цветность к.-л. цвета определяется не тремя его ЦК, а соотношением между ними, т. е. положением в ЦП прямой, проведённой из начала координат через точку данного цвета. Другими словами, цветность определяется только направлением цветового вектора, а не абс. его величиной и, следовательно, её можно охарактеризовать положением точки пересечения этого вектора с единичной плоскостью. Вместо треугольника Максвелла часто используют цветовой треугольник более удобной формы — прямоуг. и равнобедренный. Положение точки цветности в нём определяется двумя координатами цветности, каждая из к-рых равна частному от деления одной из ЦК на сумму всех трёх ЦК. Двух координат цветности достаточно, т. к., по определению, сумма её трёх координат равна 1. Точка цветности опорного цвета, для к-рой три координаты равны между собой (каждая равна 1/3), находится в центре тяжести цветового треугольника.
Представление цвета с помощью ЦКС должно отражать св-ва цветового зрения человека. Поэтому предполагается, что в основе всех ЦКС лежит т. н. физиологическая ЦКС. Эта система определяется тремя ф-циями спектральной чувствительности (СЧ) трёх разл. типов приёмников света (наз. колбочками), к-рые расположены в сетчатке глаза человека и реакции к-рых, согласно наиболее употребительной трёхкомпонентной теории цветового зрения, ответственны за человеческое цветовосприятие. Реакции этих приёмников на излучение считаются ЦК в физиол. ЦКС, но ф-ции СЧ глаза не удаётся установить прямыми измерениями. Их определяют косвенным путём и не используют непосредственно в кач-ве основы построения колориметрич. систем.
Смешение цветов; кривые сложения. Св-ва цветового зрения учитываются в К. по результатам экспериментов со смешением цветов. В таких экспериментах выполняется зрит. уравнивание чистых спектральных цветов одинаковой интенсивности (соответствующих монохроматическому свету с разл. длинами волн) со смесями трёх ОЦ. Оба цвета (чистый спектральный и смесь) наблюдают рядом на двух половинках фотометрич. поля сравнения. По достижении уравнивания измеряются кол-ва трёх ОЦ и их отношения к единичным кол-вам ОЦ. Полученные величины явл. ЦК уравниваемого цвета в ЦКС. Если единичные кол-ва красного, зелёного и синего ОЦ обозначить как (К), (3), (С), а их кол-ва в смеси (ЦК) — к', з', с', то результат уравнивания можно записать в виде цветового ур-ния: Ц*=к'(К)+з'(З)+с'(С). Описанная процедура не позволяет уравнять большинство чистыхспектр. цветов со смесями трёх ОЦ прибора. В таких случаях нек-рое кол-во одного из ОЦ (или даже двух) добавляют к уравниваемому цвету. Цвет получаемой смеси уравнивают со смесью оставшихся двух ОЦ прибора (или с одним). В цветовом ур-нии это формально учитывают переносом соответствующего члена из левой части в правую. Так, если в поле измеряемого цвета был добавлен красный цвет, то Ц*=-к'(К)+з'(З)+с'(С). При допущении отрицат. значений ЦК уже все спектр. цвета можно выразить через выбранную тройку ОЦ. При усреднении результатов подобной процедуры для неск. наблюдателей получают усреднённые значения кол-в трёх ОЦ (удельные координаты Ц), смесь к-рых зрительно неотличима от чистого спектрального цвета.
Графич. зависимости кол-в ОЦ от длины волны дают т. н. кривые сложения цветов, или кривые сложения, по к-рым можно рассчитать кол-ва ОЦ, требуемые для получения смеси, зрительно неотличимой от цвета излучения сложного спектр. состава, т. е. определить ЦК такого цвета в ЦКС. Для этого цвет сложного излучения представляют в виде суммы чистых спектр. цветов, соответствующих его монохроматич. составляющим (с учётом их интенсивности). Возможность такого представления основана на одном из опытно установленных законов смешения цветов, согласно к-рому ЦК цвета смеси равны суммам соответствующих координат смешиваемых цветов. Т. о., кривые сложения характеризуют реакции на к.-л. излучение трёх разных типов приёмников света в человеческом глазе. Очевидно, что ф-ции СЧ этих приёмников представляют собой кривые сложения в физиол. ЦКС. Каждой из бесконечно большого числа возможных ЦКС соответствует своя группа из трёх кривых сложения, причём все группы сложения связаны между собой линейными соотношениями. Следовательно, кривые сложения любой ЦКС можно считать линейными комбинациями ф-ций СЧ трёх типов приёмников человеческого глаза.
Фактически основой всех ЦКС явл. система, кривые сложения к-рой были определены экспериментально описанным выше способом. Её ОЦ явл. чистые спектр. цвета, соответствующие монохроматич. излучениям с дл. волн 700,0 (красный), 546,1 (зелёный) и 435,8 (синий) нм. Исходная (опорная) цветность — цветность равноэнергетич. белого цвета Е (т. е. цвета излучения с равномерным распределением интенсивности по всему видимому спектру). Кривые сложения этой системы, принятой Междунар. комиссией по освещению (МКО) в 1931 и известной под назв. междунар. колориметрич. системы МКО RGB (от англ., нем. red, rot — красный, green, gr?n — зелёный, blue, blau — синий, голубой), показаны на рис. 1. Кривые сложения системы МКО RGB имеют отрицат. участки (отрицат. кол-ва ОЦ) для нек-рых спектр. цветов, что неудобно при расчётах. Поэтому наряду с системой RGB МКО в 1931 приняла другую ЦКС, систему XYZ, в к-рой отсутствовали недостатки системы RGB и к-рая дала ряд возможностей упростить расчёты. ОЦ (X), (Y), (Z) системы XYZ — это нереальные цвета, выбранные так, что кривые сложения этой системы (рис. 2) не имеют отрицат. участков, а координата Y равна яркости наблюдаемого окрашенного объекта, т. к. кривая сложения у совпадает с ф-цией относительной спектральной световой эффективности стандартного наблюдателя МКО для дневного зрения.
38.
1. Основные понятия темы
Специфические реакции позволяют обнаруживать ион в отдельной порции анализируемого раствора, не считаясь с присутствием других ионов. При этом последовательность обнаружения ионов может быть произвольной.
Дробным анализом называют обнаружение ионов с помощью специфических реакций в отдельных порциях анализируемого раствора, производимое в любой последовательности.
Дробный анализ применяют агрохимические и заводские лаборатории, особенно в тех случаях, когда состав исследуемого материала достаточно хорошо известен и требуется только проверить отсутствие некоторых примесей. Если же используемые реакции не специфичны, а мешающее действие посторонних ионов устранить не удается, то проведение дробного анализа невозможно. В этом случае применяют систематический ход анализа.
Сисmемаmическим ходом анализа – называется определенная последовательность выполнения аналитических реакций, при которой каждый ион обнаруживают после того, как будут обнаружены и удалены другие ионы, мешающие его обнаружению.
Допустим, что раствор нужно испытать на присутствие катиона Са2+, но в нем одновременно может содержаться и ион Ва2+. Катион Са2+ принято обнаруживать в виде оксалата:
CaC12 + (NH4)2С2О4 = СаС2О4  + 2NH4Cl
+ 2NH4Cl
Эта реакция достаточно чувствительна, но не специфична, так как оксалат аммония (NH4)2C204 дает белый кристаллический осадок не только с Са2+, но также с Ва2+ и некоторыми другими ионами. Поэтому прежде чем обнаруживать катион Са2+, необходимо проверить, присутствует ли в растворе мешающий ион Ва2+. Последний можно обнаружить в отдельной порции раствора, действуя хроматом калия, с которым Ва2+ дает характерный желтый осадок:
BaCl2 + К2Сг04 = ВаСг04↓ + 2KCl
Присутствие иона Са2+ не мешает обнаружению иона Bа2+ этой реакцией, так как хромат кальция СаСгО4 хорошо растворим в воде (выпадает в осадок только из очень концентрированных растворов солей кальция).
Дальнейший ход анализа зависит от результата проведенного испытания. Если окажется, что ион Bа2+ отсутствует, то в другой порции раствора можно обнаруживать катион Са2+, действуя оксалатом аммония (NH4)2С2О4. Если же катион Bа2+ присутствует, то прежде чем обнаруживать Са2+, следует полностью удалить из раствора ионы Bа2+. Для этого на весь раствор действуют избытком хромата калия К2Сr04 (или дихромата калия К2Сr207), убеждаются, что ионы Bа2+ полностью осаждены в виде хромата бария BaСrО4 и, отделив осадок, беспрепятственно обнаруживают катионы Са2+.
Следовательно, в систематическом ходе анализа применяют не только реакции обнаружения отдельных ионов, но также и реакции отделения их друг от друга.
Разделение ионов чаще всего основывается на различной растворимости аналогичных солей (например, ВаСr04 и СаСrО4). Иногда в этих целях используют и различную летучесть соединений. Так, отделение катиона NH4+ от ионов Na+, К+ и Mg2+ осуществляют выпариванием раствора и прокаливанием сухого остатка. При этом непрочные соли аммония разлагаются, улетучиваются, и соединения Na+, К+ и Mg2+ освобождаются от мешающих примесей этих солей, отделяя один ион от другого, нужно внимательно следить за полнотой этого разделения, без которой результаты анализа будут ошибочными. Например, при неполном удалении иона NH4+ можно в дальнейшем „переоткрыть” К+ и Nа+, так как с реактивами на эти катионы взаимодействуют и соли аммония. Полноту удаления мешающего иона проверяют в каждом случае специальной пробой.Систематический анализ не следует противопоставлять дробному: эти методы взаимно дополняют друг друга. Каждый из них имеет свою область применения.
2. Макро-, полумикро-, микро- и ультрамикроанализ
В зависимости от количества исследуемого вещества, объема раствора и техники выполнения операций аналитические методы качественного анализа подразделяют на макро-, микро- и полумикрометоды.
1.Макрометод - наиболее старый метод химического анализа, при котором для анализа берут сравнительно большие количества вещества и реактивов: 1 г сухого вещества или 20-30 мл реактива,
2.Микрометод - при этом методе для исследования берут примерно в 100 раз меньшие количества вещества, чем в макрометоде: 5-10 мг сухого вещества или 0.2-0.3 мл раствора, При микрометоде используют высокочувствительные реакции – микрокристаллоскопические и капельные, которые проводят на предметном стекле, а о наличии определяемого вещества судят по форме кристаллов, рассматривая их под микроскопом,
3.Полумикрометод - занимает промежуточное положение между макро- и микрометодом. Для анализа берут: 50 мг сухого вещества или 0.1-1.0 мл раствора. Применяют в основном те же реакции, что и при макрометоде, но выполняют их с меньшим количеством реагирующих веществ. Этот метод анализа наиболее удобен, так как требует наименьших затрат, времени, реактивов и оборудования.
3. Техника выполнения важнейших операций в качественном анализе
При проведении качественного анализа полумикрометодом приходится выполнять множество операций, требующих определенных навыков. К таким операциям относятся: осаждение ионов, нагревание растворов, центрифугирование, выпаривание, промывание осадка и т.д.
1.Нагревание
При нагревании пробирок с большим количеством растворов на спиртовке (или горелке) жидкость может быть выброшена паром. Поэтому лучше всего нагревание вести на слабо кипящей бане.
2.Осаждение
Реакции осаждения ионов в форме труднорастворимых соединений применяются в качественном анализе с целью:
1.отделения катионов одних аналитических групп от других;
2.выделения ионов данного элемента из смеси или удаления мешающих ионов;
3.обнаружения отдельных ионов.
При проведении реакций осаждения для отделения катионов той или иной аналитической группы следует иметь в виду, что катионов этой группы может совсем не быть в исследуемом растворе, поэтому необходимо реакцию осаждения сначала проделать в отдельной пробе с 2-3 каплями анализируемого раствора. При положительном результате этой пробы следует провести осаждение ионов данной группы из большого объема исследуемого раствора.
Техника осаждения следующая: налить в пробирку несколько капель исследуемого раствора и медленно по каплям приливать к нему соответствующий реактив, перемешивая стеклянной палочкой после добавления каждой капли реактива, что ускоряет выпадение осадка. Дать осадку осесть так, чтобы раствор над осадком стал прозрачным. Далее проверить полноту осаждения, для этого прибавить еще 1 каплю реактива и, если она не вызовет помутнения прозрачного раствора над осадком, то осаждение закончено. В противном случае осаждение надо продолжить, добавив к смеси еще несколько капель реактива. Необходимо помнить, что большой избыток реактива - осадителя может вызвать частичное или полное растворение осадка в результате образования растворимых комплексных соединений. Если осаждаемое вещество может переходить в коллоидное состояние, смесь надо подогреть на водяной бане, так как нагревание способствует процессу коагуляции.
3. Центрифугирование
В качественном анализе для отделения осадка от раствора производят центрифугирование с помощью электрической центрифуги. При работе с центрифугой следует выполнять следующие правила:
1.Следить за равномерностью загрузки. Для этого следует брать одинакового размера центрифужные пробирки и наполнять их равными объемами жидкости с осадком, но не более, чем на 2/3 высоты пробирки. При работе только с одной пробиркой в противоположный патрон центрифуги нужно помещать такую же пробирку с равным объемом воды.
2.Перед включением центрифуги обязательно нужно закрыть защитный металлический кожух.
3.Включать и выключать центрифугу необходимо посредством плавного, без рывков поворота реостата.
4.Для отделения кристаллических осадков достаточно 2-х минутное цеитрифугирование; для отделения рыхлых, хлопьевидных осадков – 3-4 минутное.
Если за это время отделение осадка не произойдет, то жидкость с осадком необходимо нагреть или добавить к ней 1 каплю раствора сильного электролита, не взаимодействующего с осадком, затем вновь отцентрифугировать. В результате коагуляции коллоидов цеитрифугирование пойдет нормально.
4.Перенесение центрифугата
Если осадок, полученный при центрифугировании, очень плотный, то надосадочную жидкость (центрифугат) можно осторожно слить с осадка в другую пробирку. Если осадок рыхлый, центрифугат переносят в другую пробирку с помощью глазной пипетки, стараясь не задеть осадок. Если центрифугата над осадком очень мало, то используют капиллярную гашетку.
5.Промывание осадка
Промывание осадка проводят в том случае, если осадок должен быть подвержен дальнейшему анализу и необходимо освободиться от примесей, попавших в него из раствора. В качестве промывной жидкости используют дистиллированную воду или разбавленный раствор осадителя. К осадку в центрифужной пробирке нужно прилить около 1 мл. промывной жидкости и тщательно перемешать ее с осадком тонкой стеклянной палочкой. Затем содержимое пробирки отцентрифугировать, жидкость удалить пипеткой. Операцию промывания повторить 2-3 раза, используя свежие порции промывной жидкости.
Когда требуется промывать осадок горячей водой, нужно в пробирку с осадком прилить немного дистиллированной воды, перемешать стеклянной палочкой и нагреть на водяной бане в течение 1-2 минут. Затем отцентрифугировать и удалить центрифугат.
6.Растворение осадка
Следует помнить, что отделенный от центрифугата осадок нельзя оставлять на длительное время, так как изменяется его структура и состав, в результате уменьшается растворимость. Поэтому свежеосажденные осадки всегда растворяются легче, чем высушенные. Для растворения к осадку нужно прилить по каплям растворитель, постоянно перемешивая его стеклянной палочкой. Не следует спешить с избытком растворителя. А если осадок растворяется медленно, лучше нагреть его на водяной бане.
7. Выпаривание растворов
Выпаривание может быть частичным и полным. Частичное выпаривание проводят с целью повышения концентрации раствора. Полное выпаривание растворителя применяется при удалении летучих веществ (например, аммиака). При этом получают сухой осадок, который затем прокаливают. Выпаривание проводят в фарфоровой чашке или тигле на электрической плитке с асбестовой сеткой. Очень малые количества растворов (1-2 капли) выпаривают на стеклянной пластинке или на часовом стекле, пользуясь предварительно нагретой асбестовой сеткой. Выпаривание с выделение вредных газов и паров (НС1, НNО3, NН3 и т.д.) следует продолжить под тягой.
8.Прокаливание осадка
Прокаливание осадка в качественном анализе проводят с целью удаления органических веществ и аммонийных солей. Прокаливание ведут в той же фарфоровой чашке или тигле, откуда предварительно была выпарена жидкость, на электрической плитке. После окончания прокаливания чашку или тигель переносят с помощью тигельных щипцов на асбестовую сетку и дают остыть.
39. Электролитическая диссоциация — процесс распада электролита на ионы при его растворении или плавлении. Диссоциация в растворах[править]
Диссоциация на ионы в растворах происходит вследствие взаимодействия растворённого вещества с растворителем; по данным спектроскопических методов, это взаимодействие носит в значительной мере химический характер. Наряду с сольватирующей способностью молекул растворителя определённую роль в электролитической диссоциации играет также макроскопическое свойство растворителя — его диэлектрическая проницаемость (Схема электролитической диссоциации).
Диссоциация при плавлении[править]
Под действием высоких температур ионы кристаллической решётки начинают совершать колебания, кинетическая энергия повышается, и наступит такой момент (при температуре плавления вещества), когда она превысит энергию взаимодействия ионов. Результатом этого является распад вещества на ионы.
Классическая теория электролитической диссоциации[править]
Классическая теория электролитической диссоциации была создана С. Аррениусом и В. Оствальдом в 1887 году. Аррениус придерживался физической теории растворов, не учитывал взаимодействие электролита с водой и считал, что в растворах находятся свободные ионы. Русские химики И. А. Каблуков и В. А. Кистяковский применили для объяснения электролитической диссоциации химическую теорию растворов Д. И. Менделеева и доказали, что при растворении электролита происходит его химическое взаимодействие с водой, в результате которого электролит диссоциирует на ионы.
Классическая теория электролитической диссоциации основана на предположении о неполной диссоциации растворённого вещества, характеризуемой степенью диссоциации α, т. е. долей распавшихся молекул электролита. Динамическое равновесие между недиссоциированными молекулами и ионами описывается законом действующих масс. Например, электролитическая диссоциация бинарного электролита KA выражается уравнением типа:
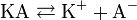
Константа диссоциации 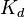 определяется активностями катионов
определяется активностями катионов  , анионов
, анионов  и недиссоциированных молекул
и недиссоциированных молекул 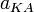 следующим образом:
следующим образом:

Значение зависит от природы растворённого вещества и растворителя, а также от температуры и может быть определено несколькими экспериментальными методами. Степень диссоциации (α) может быть рассчитана при любой концентрации электролита с помощью соотношения:
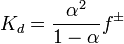 ,
,
где  — средний коэффициент активности электролита.
— средний коэффициент активности электролита.
40. Гидролиз солей.
|
|
|
|
|
Дата добавления: 2015-06-04; Просмотров: 975; Нарушение авторских прав?; Мы поможем в написании вашей работы!