КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Исследование размерных характеристик
|
|
|
|
Атомарная структура твердого тела. Кристаллическая решетка. Виды кристаллических решеток, их параметры. Индексы Миллера. Определение наночастицы. Структурные и электронные магические числа. Дефекты кристаллической структуры.
Методы изучения наноматериалов.
Лекция №6
Наноматериалы являются весьма сложными объектами для изучения. Это связано с малыми размерами структурных составляющих, спецификой многих физических свойств, большой протяженностью границ и поверхности раздела фаз, присутствием разупорядоченных и аморфных составляющих, формированием неизвестных до сих пор фаз, высокой реакционной способностью и т.д. В нанопорошках наблюдается также сильное агрегирование частиц. В связи с этим многие методы изучения крупнокристаллических материалов не применимы для наноразмерных систем, а ряд из них требуют существенных изменений и доработки.
Определение удельной поверхности нанопорошков. Для определения удельной поверхности традиционных порошковых материалов в настоящее время чаще всего используются методы измерения газопроницаемости и адсорбции.
Измерение проницаемости вещества основано на оценке фильтрационных процессов при прохождении газа через слой материала с известной пористостью. Сущность метода заключается в определении объема газа, проходящего через единицу площади за единицу времени при определенной разности давления и постоянной температуре. Как правило, перед исследованием порошки предварительно уплотняются. Данные методы имеют существенные ограничения по максимальному коэффициенту пористости изучаемого вещества и, вследствие этого, применимы лишь к порошкам с размером частиц более 10 мкм.
Адсорбционные методы делятся на статические и динамические.
Статические методы измерения основаны на достижении равновесия газ–твердое тело. Они требуют значительных затрат времени, обусловленных длительностью процесса установления адсорбционного равновесия. В динамических методах эксперименты проводятся при непрерывном течении газовой среды. Эти методы являются более производительными.
Методы газовой хроматографии основаны на различной адсорбции компонентов газовой смеси поверхностью адсорбента. Вообще, газовая хроматография – это метод разделения летучих компонентов, при котором подвижной фазой служит инертный газ (газ-носитель), протекающий через неподвижную фазу с большой поверхностью. В качестве подвижной фазы используют водород, гелий, азот, аргон, углекислый газ. Газ-носитель не реагирует с неподвижной фазой и разделяемыми веществами. Главным прибором для этого метода исследований является газовый хроматограф (рис. 1).
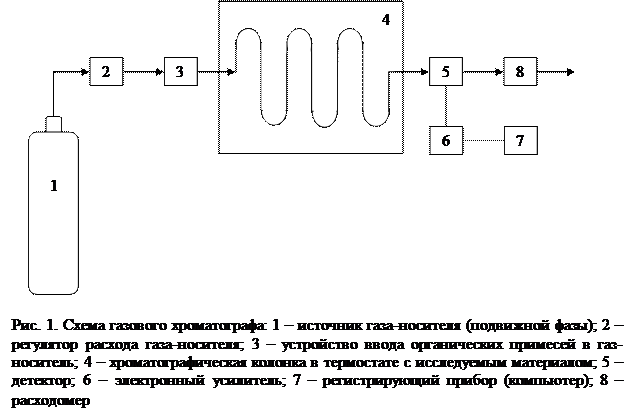
Источником газа-носителя 1, как правило, является баллон со сжатым газом, который обычно находится под большим давлением (120 атмосфер). Предназначение регулятора расхода газа-носителя 2 (редуктор, дроссель) – контроль расхода газа в системе, а также поддержка необходимого давления газа на входе в систему. Устройство ввода органических примесей в газ-носитель 3 предназначено для создания газовой адсорбционной смеси. Под хроматографической колонкой 4 подразумевается сосуд, длина которого значительно больше диаметра. Для газовой хроматографии обычно используют U-образные или спиральные колонки. Внутренний диаметр колонок – 2-15 мм, а длина – 1-20 м. Материалом для изготовления колонок служит стекло, нержавеющая сталь, медь, иногда фторопласт. В последнее время наибольшее распространение получили капиллярные колонки изготовленные из плавленого кварца, с нанесенной внутри неподвижной фазой. Длина подобных колонок может достигать сотен и даже тысяч метров, хотя чаще используются колонки длиной 30-50 м. Крайне важно плотное наполнение колонок неподвижной фазой, а также обеспечение постоянства температуры колонки в течение всего процесса хроматографирования. Точность поддержания температуры должна составлять 0,05-1 °C. Для точного регулирования и поддержания температуры используют термостаты. Детекторы 5 предназначены для непрерывного измерения концентрации органических соединений в газе-носителе на выходе из хроматографической колонки. Принцип действия детектора должен быть основан на измерении такого свойства аналитического компонента (органического соединения), которым не обладает подвижная фаза (газ-носитель).
В газовой хроматографии используют, например, такие детекторы как пламенно-ионизационный и детектор по теплопроводности (катарометр).
Схема работы пламенно-ионизационного детектора представлена на рис. 2.
Газ (A) из колонки хроматографа поступает в пламенно-ионизационный детектор. В части B поддерживается высокая температура, для того чтобы смесь оставалась в газообразном состоянии. Смешавшись с водородом (С), газ поступает в форсунку горелки детектора (Е), горение поддерживается за счет поступления кислорода (D). Пламя (F) ионизует газ, находящийся в пространстве между электродами (G, H). Ионизованные частицы уменьшают сопротивление и резко усиливают ток, который измеряется очень чувствительным амперметром. Продукты сгорания выходят через отверстие J 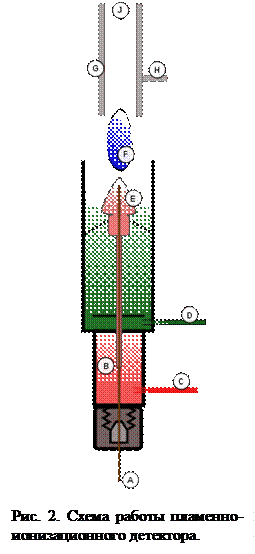 . От правильного выбора скорости потока всех используемых газов зависит стабильность и чувствительность пламенно-ионизационного детектора. Пламенно-ионизационный детектор реагирует практически на все соединения. Исключение составляют H2, инертные газы, О2, N2, оксиды азота, серы, углерода, а также воды, так как эти вещества не ионизуются под действием пламени. Величина адсорбировавшихся органических соединений определяется по изменению состава газовой смеси и, следовательно, по величине тока детектора.
. От правильного выбора скорости потока всех используемых газов зависит стабильность и чувствительность пламенно-ионизационного детектора. Пламенно-ионизационный детектор реагирует практически на все соединения. Исключение составляют H2, инертные газы, О2, N2, оксиды азота, серы, углерода, а также воды, так как эти вещества не ионизуются под действием пламени. Величина адсорбировавшихся органических соединений определяется по изменению состава газовой смеси и, следовательно, по величине тока детектора.
Более универсальным является детектор по теплопроводности (катарометр) (рис. 3). Он наиболее широко используется в газовых хроматографах. В основе его работы заложен принцип изменения сопротивления материалов от температуры.
 В полость металлического блока катарометра помещена спираль из металла с высоким термическим сопротивлением (Pt, W, их сплавы, Ni). В результате прохождения через спираль постоянного тока, она нагревается. В случае, когда спираль обмывает исходная газовая смесь, спираль теряет постоянное количество теплоты и ее температура остается постоянной. Газ, поступающий из колонки хроматографа имеет другие показатели теплопроводности, следовательно, изменяется и температура спирали. В свою очередь это приводит к изменению сопротивления нити, которое измеряют с помощью моста Уинстона. Сравнительный поток исходного газа омывает нити ячеек R2 и R4, а газ, поступающий из колонки хроматографа, омывает нити измерительных ячеек R1 и R3. Мост будет находиться в равновесии, если у четырех нитей будет одинаковой температура (одинаковое сопротивление). Если изменить состав газа, выходящего из колонки хроматографа, то сопротивление нитей ячеек R1 и R3 изменяется, равновесие нарушается и генерируется выходной сигнал.
В полость металлического блока катарометра помещена спираль из металла с высоким термическим сопротивлением (Pt, W, их сплавы, Ni). В результате прохождения через спираль постоянного тока, она нагревается. В случае, когда спираль обмывает исходная газовая смесь, спираль теряет постоянное количество теплоты и ее температура остается постоянной. Газ, поступающий из колонки хроматографа имеет другие показатели теплопроводности, следовательно, изменяется и температура спирали. В свою очередь это приводит к изменению сопротивления нити, которое измеряют с помощью моста Уинстона. Сравнительный поток исходного газа омывает нити ячеек R2 и R4, а газ, поступающий из колонки хроматографа, омывает нити измерительных ячеек R1 и R3. Мост будет находиться в равновесии, если у четырех нитей будет одинаковой температура (одинаковое сопротивление). Если изменить состав газа, выходящего из колонки хроматографа, то сопротивление нитей ячеек R1 и R3 изменяется, равновесие нарушается и генерируется выходной сигнал.
Динамический метод тепловой десорбции газа заключается в следующем. Контролируемый материал помещается в специальную ампулу (адсорбер) и непрерывно продувается смесью газа-носителя (гелия) и газа-адсорбата (азота или аргона), а на выходе адсорбера размещается детектор состава газовой смеси. Адсорбцию проводят достаточно длительное время, охлаждая материал до температуры жидкого азота, до ее полного завершения. Затем быстро нагревают адсорбер до температуры выше 200 К. При этом газ, адсорбированный на образце, быстро десорбируется. В ходе десорбции концентрация адсорбата в выходящем из адсорбера потоке газа вначале возрастает, а затем падает до прежнего значения, что фиксируется детектором в виде десорбционного пика. Площадь пика пропорциональна объему адсорбированного на образце газа. Зная этот объем и размеры молекул газа (их кинетические диаметры), мы можем вычислить площадь поверхности образца. Метод применяют для измерения удельных поверхностей в диапазоне 0,01 – 1000 м2/г при минимальной суммарной величине поверхности навески 0,05 м2. Время одиночного измерения 17–25 мин.
Для изучения наноматериалов чаще используются статические методы, схема измерений в которых заключается в следующем: дисперсное тело помещается в замкнутое пространство, заполненное газом при некотором давлении. Для наносистем применяют обычно азот или аргон. Материал начинает адсорбировать газ, что выражается в постепенном уменьшении давления газа и в возрастании массы твердого тела. После установления равновесия давление станет постоянным, а масса перестанет увеличиваться. По количеству адсорбированного газа и площади, занимаемой каждой молекулой, можно рассчитать площадь, покрытую газом-адсорбатом. Это и есть площадь поверхности исследуемого материала. Статические методы, в основе которых лежит измерение давления газа-адсорбата, называются манометрическими. Методы, основанные на взвешивании – гравиметрическими.
Удельная поверхность Sуд твердых тел может быть рассчитана по следующему соотношению:
Sуд = (xmNA / q)c, (1)
где xm – количество адсорбированного вещества, содержащегося в монослое на поверхности, моль;
NA – число Авогадро, моль–1;
c – площадь, занимаемая одной молекулой газа, адсорбированного в монослое, м2;
q – навеска порошка, кг.
В свою очередь, определение количества вещества в монослое производится по различным уравнениям изотермы адсорбции, которые представляют собой зависимость количества адсорбированного вещества от давления (или концентрации) соответствующего газа при постоянной температуре.
Наиболее часто встречающаяся экспериментальная изотерма адсорбции – S-образная изотерма – представлена на рис. 4, где p – текущее давление; pS – давление насыщения. График можно условно подразделить на три области: I – область формирования монослоя; II – область полислойной адсорбции; III – область капиллярной конденсации. Для определения величины удельной поверхности используется область I, для вычисления распределения пор по радиусам – область III.
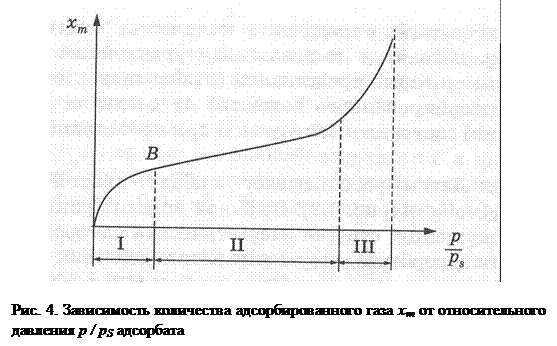
Существует несколько способов измерения количества адсорбированного в монослое вещества. В частности, для приближенной оценки можно использовать величину, которая представляет собой ординату точки перегиба изотермы адсорбции. Это приблизительно соответствует границе области, где завершается формирование монослоя. Данную точку (В) находят обычно графически (см. рис. 4). Необходимо отметить, что расчет xm требует измерений адсорбции при нескольких значениях p / pS (5-6 точек), что существенно усложняет исследование.
Определение среднего размера наночастиц. Измерение среднего размера наночастиц возможно методом малоуглового рассеяния рентгеновских лучей (МУР).
Малоугловым называется упругое рассеяние электромагнитного излучения или пучка частиц на неоднородностях вещества, размеры которых существенно превышают длину волны излучения; направление рассеянных лучей при этом лишь незначительно (на малые углы) отклоняется от направления падающего луча.
С помощью малоуглового рассеяния, в отличие от других дифракционных методов, изучают строение разупорядоченных объектов. При этом данный метод зачастую является единственным, с помощью которого можно получить информацию о хаотическом распределении неоднородностей. В структурных исследованиях вещества используют, как правило, рентгеновское излучение с длиной волны ~ от 1 до 10 Å (10 –1 – 1 нм).
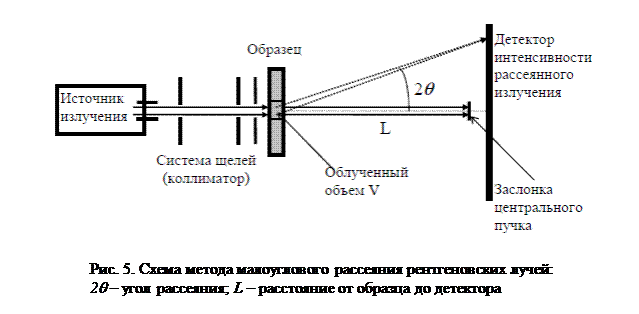
Схема метода малоуглового рассеяния рентгеновских лучей представлена на рис. 5. Источниками рентгеновского излучения служат обычные рентгеновские трубки. Для регистрации рассеянного излучения используют одноканальные ионизационные счетчики; широкое распространение получают ионизационно-чувствительные детекторы, позволяющие регистрировать одновременно всю картину малоуглового рассеяния. Tак как распределение интенсивности малоуглового рассеяния рентгеновских лучей измеряется под малыми углами, то основное требование к экспериментальной технике заключается в создании достаточно узкого нерасходящегося пучка первичного излучения. Этого достигают с помощью специальных коллимационных систем.
Дифракционная кривая, полученная в непосредственной близости от прямого пучка, позволяет установить размер и пространственное распределение наноразмерных областей неоднородности.
Процесс рассеяния можно рассматривать как интерференцию вторичных волн, излучаемых электронами вещества, колеблющимися синхронно с проходящим рентгеновским излучением. Рассмотрим интерференцию рассеянных пучков от двух точек А и B, разнесенных в пространстве на расстояние, определяемое вектором r (рис. 6).
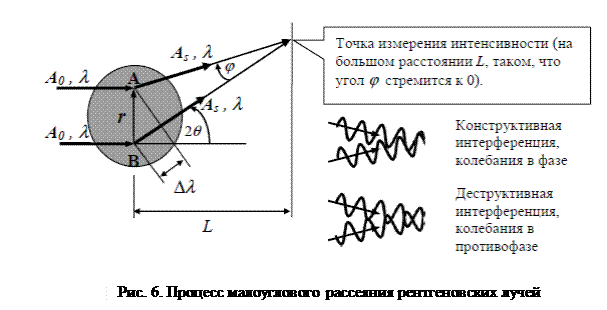
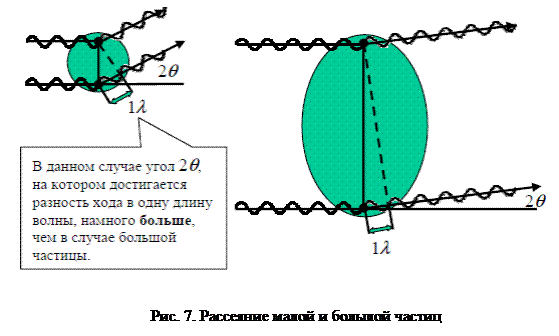
Пусть расстояние объект – детектор L таково, что направления рассеянных волн AS практически параллельны. В зависимости от величины угла 2q интерференция может приводить к удвоенной амплитуде 2 AS в точке наблюдения, если разность хода Dl кратна целому числу длин волн (суммируемые волны оказываются в фазе, конструктивная интерференция), или к нулевой амплитуде при Dl кратной половине периода волны (суммируемые волны в противофазе). Очевидно, при интерференции от множества точек максимум амплитуды будет достигаться при нулевом угле рассеяния, так как все вторичные волны приходят в этом направлении с нулевым фазовым сдвигом относительно исходной волны и относительно друг друга. С ростом угла 2q амплитуда будет спадать по некоторому закону, зависящему от пространственного расположения рассеивающих точек, то есть от структуры вещества. Таким образом, анализ зависимости амплитуды рассеянного излучения от угла рассеяния позволяет определять те или иные структурные параметры облучаемого объекта. При больших размерах рассеивающих неоднородностей кривая интенсивности будет спадать быстрее и соответственно угол 2q будет меньше, поскольку точки в таких объектах разнесены на большие расстояния и разность хода лучей в одну длину волны будет обеспечиваться при меньшем угле 2q (рис. 7).
Несмотря на большое количество достоинств, для изучения наноматериалов метод малоуглового рассеяния применяется сравнительно редко. Распространению малоугловой рентгенографии препятствуют, прежде всего, трудности обработки экспериментальных данных. Это вызвано тем, что в том случае, когда материал рассеивает рентгеновское излучение по нескольким механизмам (наличием неоднородностей электронной плотности в веществе; поверхностными несовершенствами; рассеянием на дислокациях и точечных дефектах и т.д.), не удается однозначно определить природу рассеяния. Это усложняет интерпретацию малоугловых рентгенограмм. Кроме того, затруднителен и сам расчет. Таким образом, для получения адекватных результатов необходимо иметь предельно точную кривую рассеяния, что требует специальных усовершенствований в методе и использования современных малоугловых дифрактометров.
Единственными прямыми и наиболее наглядными являются микроскопические методы определения среднего размера частиц или зерен наноматериалов. Для этого используются просвечивающие, растровые, зондовые и некоторые другие виды микроскопов.
Микроскопические методы вообще, а при изучении наноматериалов в особенности, имеют существенный недостаток – локальность, поскольку они дают информацию об очень ограниченном количестве вещества. Это важно, например, при получении корректных результатов по среднему размеру частиц. Для того чтобы набрать статистические данные, необходимо иметь большое количество микрофотографий, что чрезвычайно трудоемко ввиду локальности анализа. Если также учитывать длительность и высокую стоимость метода, то это обуславливает весьма ограниченное использование микроскопов для экспрессного анализа наноматериалов.
Несмотря на ряд ограничений, микроскопия является мощным инструментом научных исследований, единственным прямым методом изучения размерных характеристик вещества, «критерием истинности» для всех разрабатываемых косвенных методов измерения и расчета среднего размера структурных составляющих наноматериалов.
Методы измерения областей когерентного рассеяния (ОКР) (кристаллитов). Наиболее распространенными для измерения среднего размера ОКР являются дифракционные методы: рентгено- и нейтронография.
Дифракция рентгеновских лучей в настоящее время очень широко используется для исследования наноматериалов.
Рентгеновские лучи – это такие же электромагнитные волны, как и видимый свет, но со значительно более короткими длинами волн. При рентгенографических методах исследования строения твердых тел применяются рентгеновские лучи с длинами волн от 0,2Å до ~ 2,5Å, то есть примерно в 104 раз более короткими, чем длины волн видимого света. Энергия же рентгеновских квантов E=hv (v=c/l – частота; l – длина волны; c – скорость электромагнитных волн) во столько же раз выше. В силу этого рентгеновские лучи обладают уникальным свойством: они проходят через вещества, непрозрачные для видимого света. По характеру проникающей способности рентгеновские лучи делятся на мягкие (большие l – малая проникающая способность) и жесткие (малые l – большая проникающая способность). Для получения рентгеновского излучения используют установку, основной частью которой является рентгеновская трубка.
В каждом кристалле можно выделить несколько совокупностей периодически расположенных атомных плоскостей. На рис. 8, а, приведены две совокупности таких плоскостей. Рентгеновские лучи проникают внутрь кристалла и отражаются от каждой горизонтальной плоскости. В таком случае мы получаем множество когерентных пучков рентгеновских лучей, которые интерферируют между собой.
Рассмотрим два соседних пучка 1 и 2 (рис. 8, б), падающих под углом скольжения θ (угол между направлением падающих лучей и кристаллографической плоскостью). Падающий пучок возбуждает атомы кристаллической решетки, которые становятся источниками вторичных волн. Разность хода между лучами 1΄ и 2΄, отраженными от двух параллельных кристаллографических плоскостей, следующая:
АВ + ВС = 2 d sin θ,
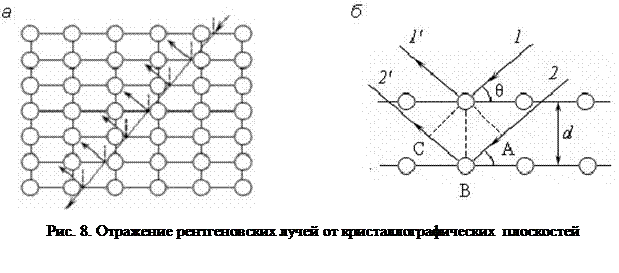 где d – расстояние между плоскостями.
где d – расстояние между плоскостями.
Максимумы интенсивности наблюдаются в направлениях, удовлетворяющих условию Вульфа-Брегга:
2 d sin q = ± n λ,
где n – целое число, называемое порядком отражения.
 Каждая совокупность периодически расположенных плоскостей дает свою систему пятен. Расположение пятен на фотопленке (рис. 9) полностью определяется расстоянием между плоскостями d. Анализируя общую картину пятен-максимумов, можно найти несколько значений d: d1, d2,... По этой совокупности параметров, можно установить тип кристаллической решетки и определить для нее расстояния между атомами.
Каждая совокупность периодически расположенных плоскостей дает свою систему пятен. Расположение пятен на фотопленке (рис. 9) полностью определяется расстоянием между плоскостями d. Анализируя общую картину пятен-максимумов, можно найти несколько значений d: d1, d2,... По этой совокупности параметров, можно установить тип кристаллической решетки и определить для нее расстояния между атомами.
Уточним смысл условия Вульфа-Брегга. Луч, который при выводе и интерпретации условия Вульфа-Брегга принято называть лучом, отраженным отдельной атомной плоскостью, в действительности не является таковым. Он возникает в результате сложного процесса, в котором участвуют атомы всего кристалла, а не только рассматриваемой атомной плоскости. Однако окончательная дифракционная картина будет такой, как если бы отдельные атомные плоскости только зеркально отражали рентгеновские лучи и не давали никаких боковых пучков.
Итак, из сказанного ясно, что если пучок рентгеновских лучей направить на кристалл, то в пространстве вокруг кристалла можно обнаружить закономерную интерференционную картину, которую сравнительно легко зарегистрировать, например, на рентгеновской пленке, помещенной на небольшом (порядка нескольких сантиметров) расстоянии от кристалла. По полученной на рентгенограмме дифракционной картине можно определять строение кристаллической решетки.
Метод дифракции нейтронов обладает существенным отличием от дифракции рентгеновского излучения. Это отличие обусловлено разной природой рассеивающих центров. Если электромагнитные волны рассеиваются электронными оболочками атомов, то нейтроны, не имеющие заряда, рассеиваются ядрами. Это приводит к тому, что дифракция нейтронов слабо зависит от атомного номера кристалла. Особый характер взаимодействия нейтронов с ядрами приводит к тому, что атомная амплитуда рассеяния нейтронов для различных элементов (в отличие от рентгеновских лучей) несистематическим образом зависит от порядкового номера элемента в периодической системе. В частности, рассеивающие способности легких и тяжелых элементов оказываются одного порядка. Поэтому изучение атомной структуры соединений легких элементов с тяжелыми является специфической областью структурной нейтронографии.
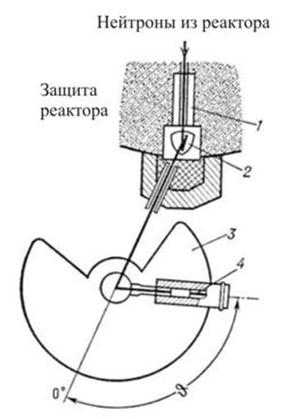
Нейтронографический эксперимент осуществляется на пучках нейтронов, получаемых в ядерных реакторах. На рис. 10 приведена типичная установка для нейтронографических исследований.
|
Нейтронографическая аппаратура (дифрактометры, нейтронные спектрометры разных типов и т.д.) размещается в непосредственной близости от реактора на пути нейтронных пучков. Плотность потока нейтронов в пучках самых мощных реакторов на несколько порядков меньше плотности потока квантов рентгеновской трубки, поэтому используемые в нейтронографии образцы существенно крупнее, чем в рентгенографии. Эксперименты могут проводиться в широком интервале температур (от 1 до 1500 К и выше), давлений, магнитных полей и т.д.
На рис. 11 приведена нейтронограмма (зависимость интенсивности рассеяния J нейтронов от угла рассеяния θ) поликристаллического образца BiFeO3. Нейтронограмма представляет собой совокупность максимумов когерентного ядерного рассеяния на фоне диффузного (некогерентного) рассеяния.
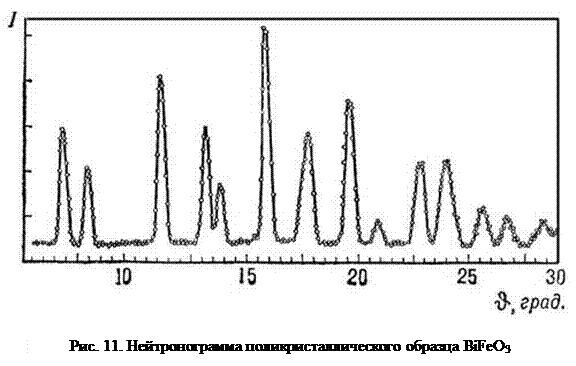
В дифракционных методах определение среднего размера ОКР основано на измерении уширения максимумов интенсивности. Регистрируемое совершенным прибором отражение от идеального монокристалла должно представлять собой вертикальную линию. В реальных системах пики интенсивности имеют определенную ширину, обусловленную как аппаратурным несовершенством, так и неидеальностью исследуемых материалов.
Методы распределения наночастиц по размерам. В настоящее время для наноматериалов распределение частиц по размерам определяют с помощью электронной микроскопии и рентгеноструктурного анализа.
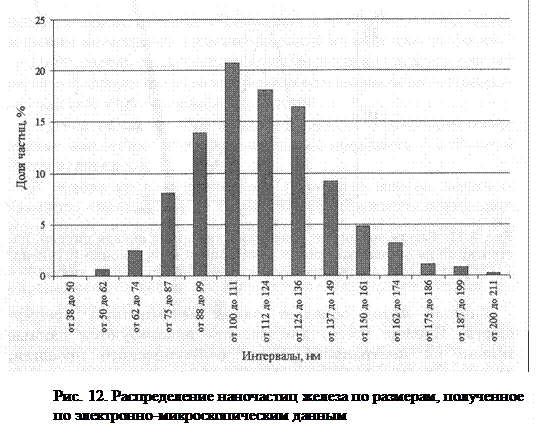
По результатам электронно-микроскопических исследований были получены первые гистограммы распределения наночастиц по размерам, для построения которых проводят измерения частиц или зерен на микрофотографиях (рис. 12). Метод очень трудоемок: чтобы получить точность эксперимента 3%, необходимо измерить диаметры более 1000 частиц. Кроме того, точность эксперимента зависит также от погрешности измерительных приборов.
Распределение наночастиц по размерам возможно на основе данных рентгеновской дифрактометрии. Однако этот способ требует тщательного проведения эксперимента в широком интервале углов дифракции и большого объема расчетов, вместе с тем точность этого способа невысока.
|
|
|
|
Дата добавления: 2014-01-03; Просмотров: 4011; Нарушение авторских прав?; Мы поможем в написании вашей работы!