КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Основы квантовой механики и строение атома
|
|
|
|
Строение вещества
I. Элементарные сведения о корпускулах и волнах и предпосылки квантовой теории. Движения корпускул и сплошных сред. Корпускулярные и волновые свойства света. Волновые и корпускулярные свойства материи. Волны материи (волны де Бройля). Простейшие виды движения частиц. Линейное движение на ограниченном интервале и модель потенциального ящика. Квантование энергии и энергетическая диаграмма. Понятие о спектральных переходах в квантовых системах. Длина волны, волновое число, частота и энергия спектрального перехода.
Основные формулы: l=h/p; l=h/mc (для электромагнитной волны);
замена с=v Þ l=h/mv (волна материи - волна де Бройля);
1) Частица в одномерном ящике
(аналогия со стоячей волной, образуемой натянутой струной)
L=nl/2, " nÎN {1, 2, 3,... ¥}; U(x) =0, E=T=p2/2m, " xÎ [0, L]
Уровни энергии частицы в одномерном “потенциальном ящике”:
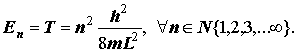
II. Движение на круговой орбите (Для самостоятельного ознакомления). Стоячие волны де Бройля на орбите и квантование величины L=mvr. Квантование классического "радиуса орбиты". Боровский радиус a0=? 2/me2. Теорема вириала и вывод формулы квантования орбитальной энергии атома H и водородоподобного иона (формула Бора). Система атомных единиц.
2) Движение электрона на круговой орбите.
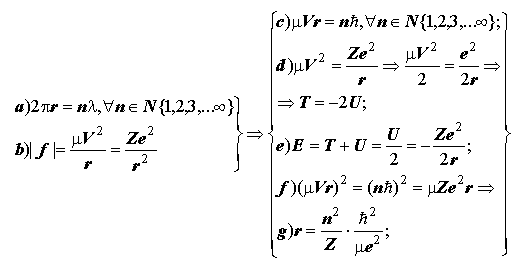
|
Уровни водородоподобного атома (иона) и радиусы орбит:
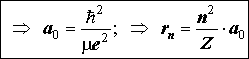
Отсюда следует формула Бора: 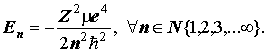
Атомная система единиц:
1) единица массы-масса электрона [M] =1 а. е. M =me;
2) единица заряда – элементарный заряд - заряд электрона [Q] =1 а. е. Q =e;
3) единица длины – боровский радиус [L] =1 а. е. L =a0;
4) В атомной системе модуль циклической константы Планка равен единице: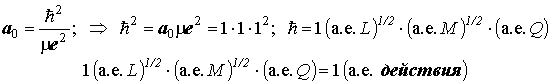
В атомной системе единиц формулы для уровней энергии и “радиусов” движения в водородоподобных атомах (одноэлектронных ионах) выглядят особенно просто:
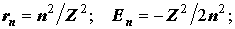
III. Уравнение плоской бегущей волны де Бройля и способ построения операторов импульса и энергии. Операторные уравнения.
3) Плоская световая волна (элекромагнитное поле):
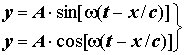 или y= A. exp [± i (wt - wx/c)]
или y= A. exp [± i (wt - wx/c)]
4) Подстановки E = ћw = mc2 Þ w = mc2/ћ = pc/ћ Þ w = E/ћ приводят к формуле для плоской волны материи 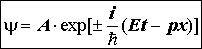
5) Плоская волна материи. Операторы динамических переменных
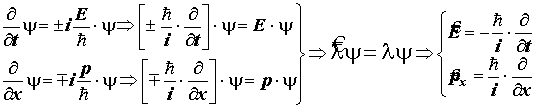
Получены важные формулы для операторов энергии и импульса
IV. Физические, математические основы, и постулаты квантовой механики. Понятие о конфигурационном пространстве (КФ) системы частиц. При описании механических движений в системе частиц {1, 2, 3,... n} используются различные пространственные переменные. Их совокупность называется конфигурационным пространством. Координаты могут быть декартовы {x1, y1, z1, x2, y2, z2, x3, y3, z3,... xn, yn, zn}, или полярные, например, шаровые {r1, J1, j1, r2, J2, j2, r3, J3, j3,... rn, Jn, jn}, или иные: {q1, q2, q3,... q‑2, q-1, qn}.
Максимальная размерность КФ 3n. В общем случае КФ является математической абстракцией. Лишь в случае одной частицы имеет геометрический смысл. Содержание постулатов квантовой механики:
Постулат 1. Волновая функция и ее свойства (конечность, однозначность, непрерывность и нормировка): y(q1, q2,... qn, t). Û òòò... òòy(q1, q2,... qn) y*(q1, q2,... qn) dv(q1, q2,... qn) =1.
Область интегрирования охватывает полный возможный диапазон значений каждой переменной. Вероятностный смысл волновой функции:
y(q1, q2,... qn) y*(q1, q2,... qn) = |y(q1, q2,... qn) |2=r(q1, q2,... qn)
|y(q1, q2,... qn) | 2dv(q1, q2,... qn) =dw(q1, q2,... qn). Кратко: |y|2dv=dw
Волновая функция (ВФ) это математический образ состояния системы – функция состояния.
Её квадрат это плотность вероятности распределения по конфигурационному пространству системы, пребывающей в некотором состоянии, которому отвечает ВФ y.
Постулат 2. Измерения физических величин и операторные уравнения. Уравнения { 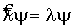 } - математические образы измерений. Операторы - образы макроскопических приборов. Связь операторов различных динамических переменных. Операторы основных динамических переменных (импульса и его компоненты, координат и потенциальной энергии, момента импульса и его компонент, кинетической энергии,). Гамильтониан.
} - математические образы измерений. Операторы - образы макроскопических приборов. Связь операторов различных динамических переменных. Операторы основных динамических переменных (импульса и его компоненты, координат и потенциальной энергии, момента импульса и его компонент, кинетической энергии,). Гамильтониан.
Постулат 3. Временное и стационарное уравнения Шрёдингера. Стационарные системы. Гамильтониан, не зависящий от времени. Основа теоретической химии - стационарное уравнение Шрёдингера.
{ 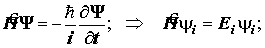 }.
}.
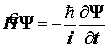 . Если гамильтониан независим от времени:
. Если гамильтониан независим от времени: 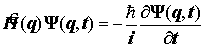 .
.
Для самостоятельного ознакомления: Стационарные системы. Подстановка с целью
разделения времени и пространственных переменных: Y(q, t) =y(q). t(t).
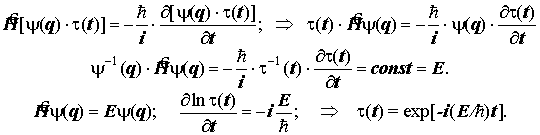
Разделение переменных приводит к двум дифференциальным уравнениям:
Пространственная часть волновой функции - стационарное уравнение Шрёдингера - это операторное выражение закона сохранения энергии в стационарной системе.
Временная часть волновой функции описывает периодический процесс. В стационарной системе все движения строго периодичны - движение постоянно повторяется с круговой частотой 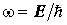 :
:
Постулат 4. Суперпозиция состояний. Состояния чистые и смешанные. Математические и физические основания принципа суперпозиции.
Формулировка:
Если две волновые функции являются решениями операторного уравнения на собственные значения, то их линейная комбинация также является решением этого уравнения.
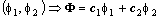
Этот принцип называется принципом суперпозиции состояний и допускает обобщение на любое число собственных функций, образующих спектр оператора.
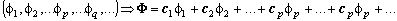
При описании состояний реальных систем в общем случае всегда возникает проблема определения коэффициентов 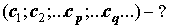
Постулат 5. Средние значения динамических переменных: 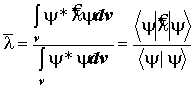
Его формулировка:
Среднее значение динамической величины, полученное в результате многих измерений, равно математическому ожиданию этой величины, вычисленному с помощью её динамического оператора.
Это утверждение на первый взгляд кажется простым следствием второго постулата, но это справедливо лишь для состояния “чистого”, волновая функция которого есть одна из простейших в спектре собственных функций динамического оператора. Для “смешанного” состояния волновая функция является уже суперпозицией более простых волновых функций, и этот постулат вводится как основание для вычислений усреднённых значений физических характеристик системы.
Для подавляющего большинства реальных систем уравнение Шрёдингера имеет слишком сложный вид, и невозможно получить спектры его собственных волновых функций и собственных значений гамильтониана (энергетических уровней всех квантовых состояний) в аналитической форме в зависимости от квантовых чисел (номеров состояний-уровней).
В силу этого расчёт свойств реальной системы почти всегда начинается с составления приближённой волновой функции для какого-то отдельно выбранного квантового состояния, а данный постулат предписывает способ вычисления наблюдаемой физической величины с помощью искусственно конструируемой волновой функции.
В этом и состоит значение 5-го постулата.
V. Уравнение Шрёдингера для простейших квантовомеханических систем.
Общая схема и примеры составления и решения уравнения Шрёдингера.
1. Одномерный "потенциальный ящик" как простейшая модель замкнутого поступательного движения. Волновые функции, граничные условия и квантование энергии (энергетический спектр). Энергетическая диаграмма и графики волновых функций. Узлы и пучности волновых функций. Нормировка. Связь номера уровня с числом узлов и пучностей волновой функции - стоячей волны де Бройля. Области применения модели "потенциального ящика".
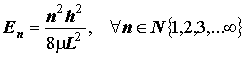
2. Понятие о трёхмерном "потенциальном ящике" как простейшей модели замкнутого пространственного движения частицы. Квантовые числа (nx; ny; nz). Уровни кубического ящика, их вырождение:
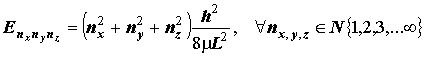
3. Плоский ротатор - простейшая модель вращения в плоскости. Условие однозначности и комплексные волновые функции плоского ротатора. Квантование энергии. Вырождение уровней. Действительные орбитали, их полярные графики и классификация состояний-уровней: {s, p, d,... }.
Рабочие формулы: Формула оператора момента импульса в плоском вращении подобна формуле оператора импульса в поступательном движении. Необходимы замены величин xÛj и m=I:
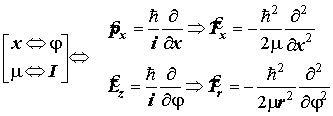 Þ
Þ
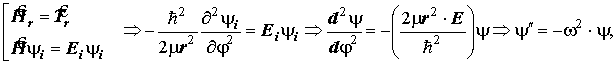
где 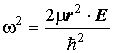
Волновые функции имеют вид: y(j) =А. exp(±iwj), Нормировка даёт А=(2p) - 1/2
Однозначность волновых функций приводит к квантованию энергии Е:
y(j) =y(j+2p) Þ exp(±iwj) =exp [±iw(j+2p)] Þ exp(±iwj) = exp(±iwj). exp(±iw2p) Þ
1= exp(±iw2p) = exp(±im2p). Отсюда w=m, а также cos(m2p) +isin(m2p) =1,
что означает cos(m2p) =1; isin(m2p) =0 Þ mÎZ0{0; ±1; ±2;... } 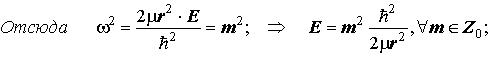
4. Гамильтониан одномерного гармонического осциллятора (Для самостоя-тельного ознакомления):
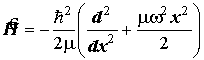 и квантование уровней колебательной энергии:
и квантование уровней колебательной энергии:
Еv=(v+1/2) hn =(v+1/2)? w " vÎN{1, 2, 3,... }.
Понятие о характеристичности колебаний химических связей и аналитические применения колебательной спектроскопии. Диаграмма энергетических уровней и графики волновых функций. Качественное сравнение волновых функций одномерного ящика и осциллятора, общие признаки, сходство и отличие.
VI. Соотношения неопределенностей Гейзенберга (Для самостоятельного ознаком-ления): Сопряженные динамические переменные (импульс-координата; энергия-время; момент импульса-угол поворота). Квант действия. Принцип исключения для совместного измерения сопряженных динамических переменных. Соотношения Гейзенберга:
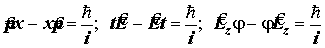 .
.
Cоотношения неопределённостей Гейзенберга относятся к числу фундаментальных законов природы. В элементарной квантовой теории их представлют также в виде произведений предельных ошибок, неизбежных при совместных измерениях, а именно:
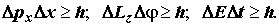
Часто соотношения Гейзенберга записывают через квадратичные отклонения в виде
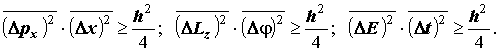
VII. Атом водорода и водородоподобные ионы в квантовой механике.
Шаровые координаты (r, J, j). Одноэлектронный гамильтониан в шаровых координатах. Уравнение Шрёдингера для водородоподобного атома. Схема разделения переменных. Атомные орбитали, их радиальные и угловые компоненты: yn, l, m(r, J, j) = Rn, l(r). ql, m(J). Fm(j). Квантовые числа n, l, m, их взаимосвязь, пределы изменения и физический смысл. Квантование энергии, модуля и проекций момента импульса. Полярные диаграммы угловых компонент АО.
Рабочие формулы: Лапласиан и его слагаемые в декартовых и шаровых координатах:
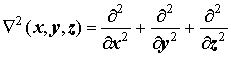
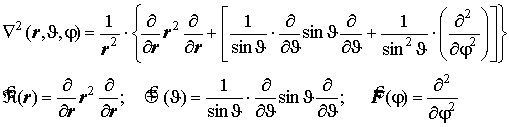
угловая часть лапласиана - оператор Лежандра: 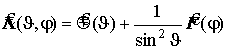
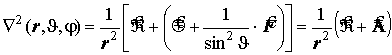 .
.
Оператор Лежандра с точностью до постоянного множителя совпадает с оператором квадрата момента импульса, а именно 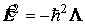
Уравнения Лапласа и Лежандра для шаровой системы (очень полезная информация):
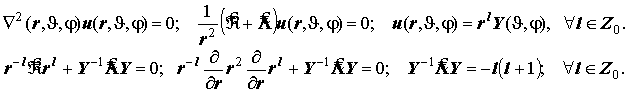
Отсюда - правило квантования модуля момента импульса при сферическом вращении.
Квантование модуля и проекций момента импульса при пространственном вращении
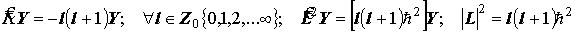
Вращательные соcтояния {s, p, d, f,... } Û l={0,1,2,3}. Углы прецессии момента импульса.
Уравнение Шрёдингера для одноэлектронного атома (водородоподобного иона). Разделение переменных и квантование динамических величин:
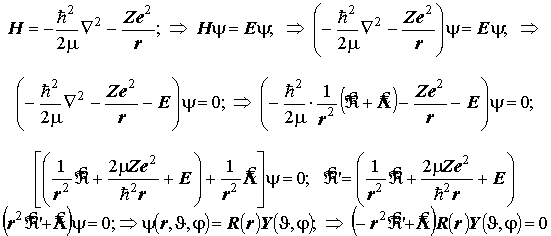
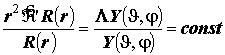
Графики радиальных компонент АО атома H и водородоподобного иона.
Радиальные распределения плотности вероятности и физический смысл боровского радиуса в квантовой механике. Энергетическая диаграмма орбитальных уровней атома водорода и водородоподобного иона и природа высокой кратности вырождения одноэлектронных (орбитальных) уровней атома
VIII. Многоэлектронный атом. Многоэлектронный гамильтониан для атома. Потенциальная энергия отталкивания электронов и ее приближенное представление в виде функции экранирования ядра. Межэлектронное отталкивание как возмущение одноэлектронного кулоновского потенциала в атоме (эффект экранирования ядра) и расщепление уровней по побочному квантовому числу l. Энергетические уровни АО многоэлектронного атома (правило Клечковского-Маделунга): “Уровни АО многоэлектронного атома возрастают с ростом суммы квантовых чисел (n+l), а при равных значениях (n+l) ниже лежит уровень с меньшим n”. Экранирование ядра. Одноэлектронный подход к проблеме строения многоэлектронного атома.
| n+l | N,l | АО | n+l | n,l | АО | n+l | n,l | АО | n+l | n,l | АО | n+l | n,l | АО | n+l | n,l | АО |
| 1,0 | 1s | 2,1 | 2p | 3,2 | 3d | 4,2 | 4d | 4,3 | 4f | 5,3 | 5f | ||||||
| 2,0 | 2s | 3,0 | 3s | 4,1 | 4p | 5,1 | 5p | 5,2 | 5d | 6,2 | 6d | ||||||
| 3,1 | 3p | 5,0 | 5s | 6,0 | 6s | 6,1 | 6p | 7,1 | 7p | ||||||||
| 4,0 | 4s | 7,0 | 7s | 8,0 | 8s |
Последовательность уровней АО многоэлектронного атома:
1s<2s<2p<3s<3p<4s<3d<4p<5s<4d<5p<6s<4f<5d<6p<7s<5f<6d<7p<8s
Качественное понятие о спине электрона и принцип Паули.
Принципы заполнения атомных орбиталей в основной электронной конфигурации: 1) водородоподобие (одноэлектронное приближение в атоме), 2) минимум энергии, 3) принцип Паули, 4) максимальный суммарный спин (1-е правило Хунда). Примеры основных электронных конфигураций легких атомов. Возбужденные атомные конфигурации.
Схема приближенного представления энергии электронного отталкивания в виде энергии экранирования ядра.
| Переменные | Слагаемые электростатической (кулоновской) потенциальной энергии | ||||||
| r1, J1, j1 | V1 | V12 | V13 | V14 | ... | ... | V1z |
| r2, J2, j2 | V2 | V23 | V24 | ... | ... | V2z | |
| ... | ... | ... | ... | ... | ... | ||
| ri, Ji, ji | Vi | Vij | ... | ... | Vi | ||
| rj, Jj, jj | Vj | Vji | ... | Vj | |||
| rz-1, Jz-1, jz-1 | Vz-1 | ... | Vz-1,z | ||||
| rz, Jz, jz | Vz |
Отдельные слагаемые равны Vi= –Ze2/ri; Vij=+Ze2/rij.
Полное выражение для электростатической потенциальной энергии: 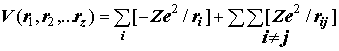
Эффективный потенциал экранирования ядра:
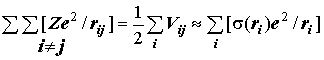
Результирующий эффективный потенциал межэлектронного отталкивания:
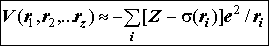
e2s(ri) - заряд экранирования (s(ri) - функция экранирования) ядра
Для одного из электронов потенциальная энергия это одно из слагаемых суммы:
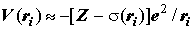
Эффективный одноэлектронный гамильтониан в многоэлектронном атоме приближённо
записывается в виде:
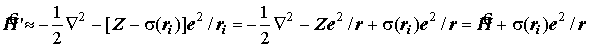
Физическим результатом “экранирования” ядра электронным облаком является дополнительное расщепление уровней АО по отношению к водородоподобному иону. Энергия АО начинает зависеть не только от главного, но и от азимутального квантового числа. Уровни АО определяются правилом Маделунга-Клечковского (см. выше).
IX. Атомные термы в приближении Рассел-Саундерса. Спин-орбитальные микросостояния атомной электронной оболочки.
Пример: первая возбужденная конфигурация атома Be(1s22s12p1), микросостояния и их качественная систематизация. Роль различных кулоновских взаимодействий: электронно-ядерного притяжения, межэлектронного отталкивания, и запрета Паули. Суммарные квантовые числа ML и MS, L и S. Атомные термы Рассел-Саундерса. Атомное внутреннее квантовое число J. Правила Хунда (первое и второе) и относительная шкала энергии атомных термов. Спектральные переходы и правила отбора (см. практические занятия). Основная конфигурация и термы атома углерода С(1s22s22p2).
X. Периодическая система Менделеева и электронные конфигурации элементов. Правило Унзольда, устойчивость сферических оболочек и природа "аномалий" у d-элементов I, VI, VIII групп Периодической системы. Качественное сравнение "сферических" электронных конфигураций некоторых d-элементов в подгруппах:
| IБ | VIБ | VIIIБ |
| 29Cu(3d104s1); | 24Cr(3d54s1); | 28Ni(3d84s2); |
| 47Ag(4d105s1); | 42Mo(4d55s1); | 46Pd(4d105s0); |
| 79Au(5d106s1); | 74W(5d46s2); | 78Pt(5d96s1); |
Термодинамика химическая
раздел физической химии (См. Физическая химия), рассматривающий термодинамические явления в области химии, а также зависимости термодинамических свойств веществ от их состава и агрегатного состояния. Т. х. тесно связана с термохимией (См.Термохимия), учением о равновесии химическом (См. Равновесие химическое) и учением о растворах (См. Растворы) (в частности, электролитов), теорией электродных потенциалов (См. Электродный потенциал), с термодинамикой поверхностных явлений.
Т. х. базируется на общих положениях и выводах термодинамики (См. Термодинамика) и прежде всего — на первом начале термодинамики (См. Первое начало термодинамики) и втором начале термодинамики (См. Второе начало термодинамики). Первое начало и важнейшее его следствие — Гесса закон служат основой термохимии. При термохимических расчётах большую роль играют теплоты образования (См. Теплота образования) веществ, значения которых для каждого из реагентов позволяют легко вычислить Тепловой эффект реакции; для органических веществ подобную роль играют теплоты сгорания (См. Теплота сгорания). Наряду с измерениями тепловых эффектов различных процессов (см. Калориметрия) используются и определение энергии связи (См. Энергия связи) между атомами на основе спектральных данных, и различные приближённые закономерности. Первое начало термодинамики лежит в основе Кирхгофа уравнения (См. Кирхгофа уравнение), выражающего температурную зависимость теплового эффекта химической реакции. Второе начало термодинамики служит основой учения о равновесии, в частности химического. Его применение к изучению химических реакции впервые было дано в работах Дж. Гиббса, А. Л. Потылицына, Г. Гельмгольца, Я. Вант-Гоффа, А. Л. Ле Шателье. В Т. х. второе начало позволяет установить, как изменение внешних условий (например, температуры, давления) влияет на равновесие и, следовательно, какими они должны быть, чтобы рассматриваемый процесс мог совершаться самопроизвольно (то есть без затраты работы извне) в нужном направлении и с оптимальными результатами.
В Т. х. для определения характеристик процесса применяют различные термодинамические функции. Наряду с энтропией (См.Энтропия) S, изменением которой наиболее просто характеризуются процессы в изолированных системах, широко используют Потенциалы термодинамические, позволяющие получить характеристики процессов при различных условиях их проведения. Так как химические реакции обычно происходят при постоянных температуре Т, давлении р или объёме V, то наибольшее практическое значение приобрели две функции:
G = H – TS, (1)
A = U – TS, (2)
где G — Гиббсова энергия, А — Гельмгольцева энергия, Н — Энтальпия и U — Внутренняя энергия. На основе (1) и (2) записываются зависимости:
Δ G = Δ H – T Δ S, (3)
Δ A = Δ U – T Δ S, (4)
где Δ Н и Δ U — соответственно изобарный и изохорный тепловые эффекты реакции. Самопроизвольные процессы, происходящие при условии р, T = const, возможны лишь в направлении уменьшения G; пределом их протекания, то есть условием равновесия, служит достижение минимального значения G. Ход процессов, происходящих при V, Т = const, прослеживается по изменению А. Знак и величина Δ G (Δ А) определяются соотношением между членами уравнения (3) или (4): тепловым эффектом Δ Н (Δ U) и так называемым энтропийным фактором T Δ S; относительное значение первого возрастает с понижением температуры, для второго — с её повышением.
В Т. х. важна роль химических потенциалов (См. Химический потенциал), так как любой переход вещества из одной фазы в другую (например, при растворении) возможен лишь в направлении их выравнивания. Условием равновесия служат одинаковые значения химического потенциала каждого компонента во всех фазах системы. Из этих условий выводится Фаз правило, являющееся фундаментальным обобщением, описывающим равновесие в любой гетерогенной системе. В Т. х. большое значение имеют различные соотношения, выводимые из общих положений термодинамики. К их числу относятся: Действующих масс закон; уравнение изотермы реакции, характеризующее зависимость Δ G (Δ A) от концентраций ( активностей (См. Активность)) и парциальных давлений (фугитивностей (См.Фугитивность)) реагентов и выражающее величину максимальной работы реакции; уравнение изобары (изохоры) реакции, характеризующее влияние температуры на химическое равновесие, и т. д.
Для расчётов равновесий существенное значение имеют так называемые Стандартные состояния веществ. Если все реагенты находятся в этих состояниях, то справедливо соотношение
Δ G 0 = –RT ln K, (5)
где G 0 — стандартная гиббсова энергия, R — Газовая постоянная, К — константа равновесия; объединение (3) с (5) даёт соотношение
–RT ln K = Δ H0 – T Δ S0, (6)
позволяющее по стандартным энтропиям и теплотам образования рассчитать разнообразные равновесия (химическое взаимодействие, фазовые равновесия в одно- и многокомпонентных системах, диссоциация электролитов, в частности комплексных соединений, и т. д.). Для расчёта химических равновесий важно Третье начало термодинамики (см. также Нернста теорема). С его помощью можно найти энтропию вещества в данных условиях на основании результатов калориметрических определений — по температурной зависимости его теплоёмкости (от температур, близких к абсолютному нулю, до данной температуры), по температурам фазовых переходов (См. Температура фазового перехода) и теплотам фазовых переходов (См. Теплота фазового перехода) (в соответствующем интервале температур). Затем по значениям S каждого реагента (S прод. и S исх. — энтальпии продуктов реакции и исходных веществ) легко вычислить Δ S (∑ S прод. — Δ S исх.) для реакции.
Важное место в Т. х. принадлежит квантовомеханическим расчётам термодинамических свойств и характеристик процессов (например, теплот образования); методами статистической термодинамики можно вычислить значение различных термодинамических функций на основе спектральных данных, связывая последние со структурой молекул (см. Статистическая физика).
Из других направлений Т. х. большая роль принадлежит термодинамике растворов. Хотя общая теория растворов не разработана, однако введение понятия активности существенно облегчило использование термодинамических уравнений (при наличии соответствующих экспериментальных данных).
Выводы и методы Т. х., связанные с термохимией, учением о химическом равновесии, свойствами растворов и т. д., широко используются и в смежных отраслях знаний (физика, теплоэнергетика, геология, геохимия, биология и др.), и при решении проблем прикладного характера (химическая, нефтехимическая, металлургическая, топливная и др. отрасли промышленности), способствуя теоретическому обоснованию и практическому осуществлению проектируемых, вновь вводимых и интенсификации ранее осуществленных процессов.
С середины 20 в. получили развитие термодинамика неравновесных процессов и термодинамика высокотемпературных химических реакций.
Лит.: Курс физической химии, 2 изд., М., 1969; Еремин Е. Н., Основы химической термодинамики, М., 1974; Карапетьянц М. Х,, Химическая термодинамика, 3 изд., М., 1975; Пригожин И., Дефэ и Р., Химическая термодинамика, пер. с англ., Новосиб., 1966; Glasstone S., Thermodynamics for chemists, N, Y., 1947; Aston J., Fritz J., Thermodynamics and Statistical Thermodynamics, N. Y.—L., 1939; Lewis G., Randall М., Thermodynamics, 2 ed., N. Y. — L. — Toronto, 1961. см. также лит. при ст. Термодинамика.
М. Х. Карапетьянц.
|
|
|
|
|
Дата добавления: 2013-12-12; Просмотров: 961; Нарушение авторских прав?; Мы поможем в написании вашей работы!