КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Коллигативные свойства растворов
|
|
|
|
А В
В А
Соединение атомов происходит в результате образования химических связей между ними. Химические связи – это результат изменения состояния внешних электронов атомов, сопровождающегося понижением их энергии. Поэтому при классификации химических реакций учитывается изменение состояния электронов и характера химических связей. Все реакции оказалось возможным разделить на два типа – окислительно-восстановительные и кислотно-основные. Об окислительно-восстановительных реакциях вы имеете достаточное представление. Они характеризуются переносом разного числа электронов от одних атомов к другим и вытекающим отсюда изменением степеней окисления.
Класс явлений, изучением которых занимается химия, сводится к соединению атомов между собой, разъединению атомов и перегруппировкам атомов. Это выглядит довольно просто. Но подробное изучение этих процессов приводит к накоплению огромного фактического материала, созданию многих теорий, и все это составляет то, что называется наукой химией. С соединением атомов приходится сталкиваться сравнительно редко, главным образом в лабораторных экспериментах, потому что все они уже соединены между собой (в условиях нашего обитания). Разъединение атомов происходит, главным образом, при очень сильном повышении температуры. Таким образом, самой широко распространенной группой явлений оказываются перегруппировки атомов – то, что мы встречаем в большинстве химических реакций.
Но о реакциях второго типа обобщенного представления у вас нет. Часто вспоминают, что есть реакции замещения, обмена, соединения и разложения. Но это классификация по другому признаку – не по электронным процессам, а по числу исходных веществ и продуктов реакции. На самом деле все реакции, не являющиеся окислительно-восстановительными, можно объединить по общему признаку передачи электронных пар от одних атомов к другим, и назвать кислотно-основными. О них мы и будем говорить на трех очередных лекциях. Можно сказать, что все вещества проявляют кислотные и основные свойства, а часто и то, и другое сразу.
В чем причина того, что обширный тип реакций назван именно кислотно-основными реакциями? Кислоты представляют собой самый древний класс веществ, хотя в древности они не назывались классом. Просто они выдавали себя кислым вкусом. Способность воспринимать этот вкус является прямым указанием на особое значение кислот для живых организмов.
В развитии химии важным этапом было получение концентрированных растворов сильных кислот. С их помощью можно было растворять металлы, различные минералы, полностью переводить в раствор вещества растительных и животных клеток. Тогда же были открыты вещества, нейтрализующие кислоты, устраняющие их химическое действие и кислый вкус. Они были названы щелочами. Постепенное развитие химии вызывало возникновение все более широких обобщений относительно природы кислотно-основного взаимодействия, что и привело в итоге к разделению всех химических процессов на окислительно-восстановительные и кислотно-основные.
Проблема кислот и оснований оказалась настолько сложной, что привела к возникновению целого ряда теорий, некоторые из которых сохраняют свое значение и в настоящее время. Из старых теорий можно упомянуть кислородную теорию, выдвинутую в 1777 г. великим французским химиком А. Лавуазье. Он считал, что наиболее чистый удобовдыхаемый воздух представляет собой непременное кислотообразующее начало. По Лавуазье, этот газ («начало») входит в состав всех кислот, что и отражено в названии, предложенном Лавуазье для него – в переводе на русский язык это кислород (oxygenium). Здесь Лавуазье проявил излишнюю категоричность. Он утверждал, что кислород есть и в составе таких кислот, как хлороводород, которые позднее стали называться бескислородными. Делались попытки разложить хлороводород на кислород и особый элемент мурий. С современных позиций рациональное зерно кислородной теории Лавуазье можно видеть в том, что большинство кислот это так называемые ОН-кислоты, в которых замещающийся водород связан с атомом кислорода.
Спустя 110 лет новую и очень успешную теорию кислот и оснований выдвинул шведский химик С. Аррениус. Он объяснил природу кислот и оснований на основе своей теории электролитической диссоциации. Кислота является источником ионов водорода, Н+, а основание – источником ионов гидроксида, ОН– . Эти ионы при реакции кислоты с основанием образуют молекулы воды. Теория Аррениуса все же имеет существенные недостатки. В качестве оснований в ней рассматриваются только гидроксиды. Кислотами по этой теории не могут быть неэлектролиты. Вещества жестко делятся на классы кислот, оснований, оксидов, солей, но свойства кислотности и основности могут проявляться в разных классах веществ. Рассмотрение кислотно-основных взаимодействий ограничивается только водными растворами. И, наконец, в теории Аррениуса не могла быть учтена особая роль иона водорода (протоны и электроны еще не были известны) как частицы, по своим характеристикам отличающейся от всех других ионов. На теории Аррениуса не могло закончиться познание сущности кислотно-основных свойств веществ.
В 20-х годах XX века в результате открытия субатомных частиц (пока только электронов и протонов) и утверждения в химии теории строения атома, появляются новые теории кислот и оснований, делающие понятной сущность кислотно-основ-ных взаимодействий в различных условиях. Эти теории не противоречат одна другой, а дополняют друг друга.
Одна из теорий, появившихся в этот период, получила название теории сольво-систем. Она применима к кислотно-основным взаимодействиям в любых растворителях и позволяет рассматривать в качестве кислот и оснований вещества разных классов. Кэди и Илси (1928) предложили следующие определения кислот и оснований. Кислота – вещество, повышающее при растворении концентрацию катионов данного растворителя. Основание – вещество, повышающее концентрацию анионов растворителя. Теория охватывает любые механизмы, приводящие к изменению концентрации ионов. Если взять в качестве растворителя воду, то такие вещества, как гидроксид калия и аммиак окажутся основаниями. Но они проявят свои свойства по разным механизмам. Гидроксид калия непосредственно образует анионы растворителя OH– при растворении:
KOH = K+ + OH–
Аммиак будет обратимо реагировать с водой, так же повышая концентрацию анионов растворителя:
 NH3 + H2O NH4+ + OH–
NH3 + H2O NH4+ + OH–
В качестве примера кислоты возьмем оксид серы(VI). Известно, что при растворении в воде получается раствор с кислотными свойствами, то есть повышается концентрация катионов растворителя Н+. Кислая среда возникает вследствие образования серной кислоты по реакции
SO3 + H2O = H2SO4,
но в данном контексте важно, что непосредственно оксид серы(VI) проявляет себя как кислота.
Существенную роль в химии играет электронная теория кислот и оснований, предложенная американским химиком Гилбертом Льюисом (1926). Она с единых позиций охватывает все химические реакции, не сопровождающиеся изменением степеней окисления атомов. Обычные кислоты и основания, рассматривающиеся в теории Аррениуса, в электронной теории становятся одной из разновидностей в обширной совокупности кислот и оснований.
Кислотой в теории Льюиса считается частица, содержащая атом – акцептор электронной пары. Такая частица называется также электрофилом. Основание – частица, содержащая атом донор электронной пары. Другое название такой частицы – нуклеофил. Таким образом, в основе кислотно-основного взаимодействия оказывается способность образовывать химические связи по донорно-акцепторному механизму.
В общем виде кислоту обозначают как частицу со свободной орбиталью
 А или просто А
А или просто А
Основание обозначают как частицу с неподеленной электронной парой
В: или просто В
Свободные орбитали и неподеленные пары не обязательно имеются в основном состоянии реагентов, но могут возникать в ходе химического взаимодействия. В непредельных соединениях роль свободной электронной пары может выполнять электронная пара π-связи.
Кислотно-основные реакции могут иметь характер соединения, обмена, замещения. Представления электронной теории применимы как к неорганическим, так и к органическим веществам и реакциям. Рассмотрим пример – присоединение хлороводорода к этилену. Сначала ион водорода Н+ атакует атом углерода, притягивая наиболее подвижную пару электронов π-связи. Смещение π-электронов обозначают дугообразной стрелкой:

+

 Н Н Н
Н Н Н

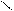 С = С + Н+ ® С – СН3
С = С + Н+ ® С – СН3
Н Н Н
На этой стадии реакции молекула этилена играет роль основания, а ион водорода – кислоты. В образовавшейся частице (карбокатионе) атом углерода имеет свободную орбиталь и становится кислотой.
На второй стадии реакции к атому углерода в карбокатионе присоединяется анион хлора, имеющий 4 неподеленные электронные пары. Карбокатион играет роль кислоты, а ион хлора – основания. Для изображения схемы взаимодействия достаточно обозначить одну электронную пару хлора:
|
 Н
Н

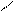 С – СН3 +:Cl– ® СН2Cl – СН3
С – СН3 +:Cl– ® СН2Cl – СН3
 Н
Н
Из показанного схематически процесса присоединения хлороводорода к этилену видно, что на первой стадии органическая молекула реагирует как основание, а на второй стадии образовавшийся карбокатион – как кислота.
Вообще, теория механизмов органических реакций базируется на электронной теории кислот и оснований.
Электронная теория Льюиса широко применяется в химии комплексных соединений, так как связи между центральным атомом и лигандами образуются по донорно-акепторному мехенизму. Центральный атом со свободными орбиталями это кислота, лиганды с неподеленными электронными парами – основания. Здесь, однако, обнаруживается слабая сторона теории Льюиса. Это невозможность построения единой шкалы для силы льюисовских кислот и оснований. Это можно показать на примере устойчивости комплексных соединений. Комплекс бериллия с ионами фтора [BeF4]2– более устойчив, чем комплекс меди [CuF4]2–. Это можно считать признаком более сильных кислотных свойств иона Be2+ по сравнению с ионом Cu2+. Но если сравнить устойчивость аммиачных комплексов, то оказывается, что [Cu(NH3)4]2+ более устойчив, чем [Be(NH3)4]2+. Очевидно, что сила кислот изменяется в различной последовательности по отношению к разным основаниям. Иначе можно сказать, что наблюдается избирательность взаимодействия кислот с разными основаниями.
Следует отметить, что представления о кислотах и основаниях в теории сольвосистем согласуются с представлениями электронной теории. Выше рассмотрен пример оксида SO3, показывающий, что он ведет себя как кислота. Выясним, является ли он кислотой согласно теории Льюиса. Представим схематически механизм реакции с водой:
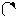 |
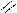
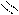
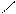
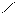 O H O O–
O H O O–
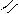
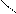
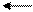 O = S: O → S + H+
O = S: O → S + H+
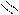 O H O OH
O H O OH
Атом серы в оксиде проявляет себя как кислота, будучи акцептором электронной пары кислорода, находящегося в молекуле воды. При этом электроны π-связи смещаются от серы к кислороду. Один из протонов воды остается в виде иона.
На электронной теории кислот и оснований основана концепция жестких и мягких кислот и оснований (ЖМКО), предложенная в 60-х годах прошлого века Р. Пирсоном. Концепция ЖМКО позволяет прогнозировать избирательное взаимодействие льюисовских кислот и оснований.
Жесткие кислоты и основания – это частицы, точнее соответствующий центр (атом) в частицах – имеющие малый радиус, трудно поляризующиеся.
Мягкие кислоты и основания – это частицы, имеющие кислотный или основной центр (атом) большого радиуса, легко поляризующийся.
| жесткие | мягкие | |
| Кислоты А | H+ (предельно жесткая), Na+, Ca2+, Mg2+, Al3+, H2O, Fe3+, | Ag+, Cu+, Tl+, Cd2+, Hg2+ |
| Основания В | F–, OH–, O2–, H2O, Cl– | I–, S2–, R–S–, As3– |
В группах сверху вниз наблюдается переход от жестких к мягким частицам (галогены).
Так в чем же значение этой классификации кислот и оснований?
Жесткие кислоты преимущественно образуют связи с жесткими основаниями, а мягкие кислоты – с мягкими основаниями.
В этом состоит предсказательное значение концепции ЖМКО.
Мягкие частицы образуют между собой преимущественно ковалентные связи, а жесткие – ионные.
Самопроизвольные кислотно-основные реакции идут не только в направлении от сильных к слабым, но и от частиц, сочетающих жесткий центр с мягким, к частицам, сочетающим мягкий центр с мягким и жесткий центр с жестким.
В организме человека жесткие кислоты Na+, K+, Mg2+, Ca2+,H+. Жесткие основания ОН –, F –
Мягкие кислоты – ионы d -элементов (медь, железо и др.). Железо(II) мягче железа(III). Мягкие основания – HS–, RS–, RSR. Токсичность ионов тяжелых металлов (таллий, ртуть, свинец) обусловлена их способностью образовывать связи с серой белков. Это ведет к блокированию ферментативной активности.
Явление симбиоза. Мягкий лиганд смягчает жесткую частицу:
KA–Zn2+ – связи с ионами галогенов ослабевают в ряду I – > Br – > Cl – > F – (КА – карбоангидраза)
H2O–Zn2+ – связи с ионами галогенов ослабевают в ряду F – > Cl – > Br – > I –
Важна не только характеристика частицы как таковой, но и ее положение относительно воды как растворителя, в котором обычно и осуществляются интересующие нас превращения. Например, среди галогенов фтор отделен от хлора в ряду понижения жесткости располагающейся между ними водой (также и ОН–):
F– > H2O (OH–) > Cl– > Br– > I–
Отсюда возникают любопытные закономерности в растворимости:
Ag+ < H2O < Li+ Li+×nH2O + F– = LiF¯ + nH2O
I– < H2O < F– Ag+×nH2O + I– = AgI¯ + nH2O
AgF + LiI = AgI¯ + LiF¯
Растворимость AgF >> AgCl > AgBr > AgI
170 г 0,002 г
Растворимость LiF << LiCl < LiBr < LiI
0,26 г 78,5 177 165
Дополнительное.
Радиус атома Н 50 пм (10–10 м). Радиус иона Н+» 0,01 пм (10–14 м).
Радиус атома Li 155 пм (10–10 м). Радиус иона Li+ 68 пм.
| Жесткие кислоты | Мягкие кислоты | ||
| +3 +4 | Сr, Co, Fe, As, CH3SH Si, Ti, Zr, U, Sn | +1 +2 | Cu, Au, Tl, Hg2+ Pd, Cd, Pt, Hg, CH3Hg+, Co(CN)52–, Pt4+, Tl3+ |
| Жесткие основания | Мягкие основания | ||
| H2O, OH–, F–, NO3–, CH3COO–, PO43–, SO42–, Cl–, CO32–, NH3, RNH2 | R2S, RSH, RS–, I–, SCN–, S2O32–, R3P, CN–, R-NC, CO, C2H4, H–, R– |
Промежуточные основания: C2H5NH2, C5H5N, NO2–, SO32–, O2, Br–.
1. В природе нет чистой воды. Есть только водные растворы. Не стану вам рассказывать, что такое растворы, но напомню определение:
Раствор это гомогенная система, состав которой можно непрерывно изменять в определенных пределах, и в которой имеется взаимодействие между частицами веществ.
Встречается ограниченная растворимость от 0 до s (B), (В – растворенное вещество, А – растворитель),
и неограниченная растворимость:
мольная доля Х(В) от 0 до 1
одновременно мольная доля Х(А) от 1 до 0
Практически особенно широко применяются жидкие растворы. В чем причины этого?
– в жидких растворах химические реакции протекают в объеме, и поэтому все растворенное вещество доступно для химического превращения;
– растворы имеют собственные физические свойства, отличные от свойств индивидуальных веществ;
– химическая активность вещества в растворе изменяется в зависимости от его концентрации;
– вещество в растворе легко дозировать.
2. Термодинамика растворения. Возможность образования раствора с достаточно высокой концентрацией вещества определяется энтальпийным и энтропийным факторами. Сравним переход хлорида калия в газообразное состояние и растворение его в воде:
Δ H º Δ S º Δ G º
кДж/моль Дж/моль·К кДж/моль
KCl(тв) → KCl(г) +214 +153 +169
KCl(тв) → KCl(р-р) +16,7 +75 –5,7
Энтропийный фактор в обоих случаях способствует процессу. Но при переходе в газообразное состояние процесс контролируется энтальпийным фактором – энергия затрачивается на разрушение кристаллической структуры. При обычной температуре процесс не идет. При образовании раствора происходит гидратация ионов соли. Выделяющаяся теплота в значительной мере компенсирует энергию кристаллической структуры, и процесс в этом случае контролируется энтропийным фактором.
При теоретическом рассмотрении растворов вводится понятие идеальных растворов – это такие растворы, в которых энергии взаимодействия между всеми видами частиц одинаковы: Е (А–А) = Е (В–В) = Е (А–В). При образовании идеальных растворов имеет место аддитивность объемов. Смесь воды и спирта явно не идеальный раствор, так как наблюдается уменьшение объема смеси V (Н2О) + V (С2Н5ОН) > V (смесь). К идеальным растворам наиболее близки смеси веществ, состоящих из разных изотопов. Например, воде содержится ряд разновидностей молекул с изотопами 2Н, 17О, 18О.
3. Растворимость газов. Все газы растворяются в жидкостях. В качестве количественной характеристики удобен объемный коэффициент растворимости KV, численно равный объему газа, растворяющегося в 1 л жидкости при данной температуре. При этом давление не влияет на KV, но влияет на растворимость газа, так как один и тот же объем содержит количество вещества газа, пропорциональное давлению. Различают плохо растворимые газы с объемными коэффициентами растворимости много меньше единицы (KV <<1, кислород KV = 0,031 при 20ºС), умеренно растворимые газы (KV не сильно отличается от 1; СО2 KV = 0,88) и хорошо растворимые газы ((KV >>1, аммиак KV = 710).
Рассмотрим закрытую систему, состоящую из жидкости А и газа В:

На границе раздела фаз устанавливается равновесие
 В(г) В(р-р)
В(г) В(р-р)
Применим ЗДМ:


Следовательно, растворимость газа в жидкости пропорциональна давлению газа над поверхностью жидкости (закон Генри).
Часто приходится рассматривать растворимость некоторого газа, содержащегося в газовой смеси. Если считать, что состояние газа близко к идеальному газу, то каждый газ растворяется независимо от других газов данной смеси, но в соответствии со своим парциальным давлением. Формула принимает следующий вид:

Это закон Генри-Дальтона: Если газ входит в состав смеси, то растворимость его в жидкости пропорциональна парциальному давлению газа над жидкостью.
Растворимость газов зависит от природы растворителя и, в частности, от присутствия в растворе электролитов. Знаменитый отечественный физиолог И. М. Сеченов, изучая физиологию дыхания, установил, что растворимость газов уменьшается при повышении концентрации электролита по следующему уравнению:

где s вода – растворимость газа в воде, s эл – растворимость в растворе электролита, с эл – концентрация электролита в данном растворе. Эта зависимость носит название закон Сеченова.
4. Коллигативные свойства растворов. Так называют свойства, которые не зависят от природы растворенного вещества, а зависят только от концентрации свободно движущихся растворенных частиц (молекул, ионов). Эти свойства называют также осмотическими.
4.1. Давление пара растворителя над растворами. Над жидкостью в замкнутом объеме образуется насыщенный пар, давление которого увеличивается при повышении температуры. Это явление всем известно по понятиям сухого и влажного воздуха. Над различными бассейнами воды нет насыщенного пара, так как нет замкнутого объема. Поэтому и наблюдается переменная влажность – водяной пар в воздухе ненасыщенный. Мы должны представить себе жидкость, над которой имеется замкнутое пространство, содержащее только пары данной жидкости.
|

В случае индивидуальной жидкости А давление насыщенного пара при данной температуре постоянно. Чем ниже температура кипения жидкости, тем выше давление пара при одинаковой температуре. Если в замкнутый объем помещен разбавленный раствор нелетучего вещества (сахариды, мочевина и др.), то над ним устанавливается несколько пониженное давление пара. Зависимость понижения давления пара от концентрации принимает наиболее простой вид, если концентрацию выражать в мольных долях Х:
 ;
;  .
.
Для разбавленных растворов выполняется соотношение, называемое первым законом Рауля:

При наличии растворенного вещества уменьшается химический потенциал жидкости (вспомните,  ), вследствие чего равновесие смещается в направлении конденсации (понижения давления пара), так как при исходном давлении пара р º(А) его химический потенциал окажется выше, чем μ(А) в растворе.
), вследствие чего равновесие смещается в направлении конденсации (понижения давления пара), так как при исходном давлении пара р º(А) его химический потенциал окажется выше, чем μ(А) в растворе.
Разность р º(А) – р (А) называется понижением давления пара жидкости над раствором, Δ р (А). Используя это понятие, зависимость можно представить следующим образом:

На основе этого уравнения закон Рауля формулируется так: Относительное понижение давления пара растворителя над раствором по сравнению с индивидуальным растворителем равно мольной доле растворенного вещества.
4.2. Понижение температуры замерзания и повышение температуры кипения растворов. Здесь нельзя не вспомнить, как в морозы тают на дорогах снег и лед при обработке «химическими реагентами». В этом и проявляется понижение температуры замерзания. Понять эти два явления проще всего, рассматривая изменение химического потенциала жидкости при повыше
нии температуры.

Согласно определению,
 ,
,
химический потенциал индивидуального вещества понижается с ростом температуры. При фазовых переходах тв→ж и ж→г энтропия растет без изменения температуры, вследствие чего наклон линий становится круче. Растворитель мы рассматриваем на температурном интервале жидкости от Т з до Т к. Когда вместо индивидуальной жидкости берется раствор, то химический потенциал жидкости понижается, и линия з - к, соответствующая химическому потенциалу раствора, проходит ниже. Равновесие раствор–твердый растворитель устанавливается при более низкой температуре, а равновесие раствор –газообразный растворитель – при более высокой температуре. Иначе говоря, у раствора понижена температура замерзания и повышена температура кипения. При повышении концентрации растворенного вещества линия раствора проходила бы ниже, что означало бы дальнейшее понижение Т з и повышение Т к. Обратите внимание: график показывает, что при прочих равных условиях Δ Т з > Δ Т к.
Согласно второму закону Рауля, понижение температуры замерзания раствора по сравнению с индивидуальным растворителем пропорционально концентрации растворенного вещества. Аналогичное утверждение справедливо и для повышения температуры кипения.
Для того, чтобы представить зависимости в наиболее простой форме, введем еще один способ выражения концентрации – моляльную концентрацию.
Моляльная концентрация b численно равна количеству растворенного вещества, приходящемуся на 1 кг (1000 г) растворителя:

Теперь можно написать уравнения для закона Рауля:
Δ Т з = K · b; 
Δ Т к = E · b; 
Коэффициенты K и E представляют собой константы растворителя, и называются, соответственно, криометрическая константа и эбулиометрическая константа. Так, для воды K = 1,86 К·кг·моль–1 и E = 0,52 К·кг·моль–1. Значениями констант подтверждается, что Δ Т з данного раствора больше, чем Δ Т к.
Из формул закона Рауля следует, что точное определение Δ Т з (Δ Т к) для раствора, приготовленного из точно известных масс растворителя и растворенного вещества, позволяет вычислить молярную массу вещества в растворе. Значения Δ Т должны быть определены с точностью до 0,01 К. Для этого сконструированы специальные термометры.
4.3. Осмос. Явление осмоса постоянно наблюдается в природе. Восстановление тонуса увядших растений при поливе водой – это результат осмоса.
Осмосом называется явление самопроизвольного переноса растворителя через полупроницаемую мембрану из чистого растворителя в раствор, или из раствора с меньшей концентрацией растворенного вещества в раствор с большей концентраций.
Под свойством полупроницаемости подразумевается, что мембрана пропускает только молекулы растворителя и не пропускает более крупные молекулы растворенного вещества. Полупроницаемыми мембранами являются клеточные мембраны (внешние и внутренние) и разнообразные искусственные пленки.
В системе с мембраной, разделяющей раствор и растворитель, диффузия растворителя через мембрану идет с большей скоростью из растворителя в раствор, так как в растворе концентрация растворителя меньше. Результатом является перенос растворителя и увеличение объема раствора. От этого возрастает тургор (напряженное состояние) клеток. Осмос демонстрируется на приборе осмометре (на рисунке показана принципиальная схема осмометра без соблюдения масштаба).

В перевернутую воронку, закрытую мембраной (пунктирная линия) помещается раствор (взят пример глюкозы). Воронку погружают в сосуд с водой. Вода, проникающая в раствор в результате осмоса, увеличивает объем раствора, и он начинает подниматься по трубке вверх. Возникает гидростатическое давление столба жидкости. При достижении некоторой высоты столба подъем жидкости прекращается, устанавливается осмотическое равновесие. Гидростатическое давление столба жидкости становится равным движущей силе осмоса, которая называется осмотическим давлением π. Осмотическое давление проявляется по мере приближения системы к равновесию. Например эритроцит, помещенный в водную среду, постепенно раздувается, мембрана натягивается. Прочность ее недостаточна, чтобы выдержать равновесное осмотическое давление, и в какой-то момент клетка лопается. Происходит гемолиз.
Явления, наблюдаемые в осмометре, наглядно подтверждают, что за счет самопроизвольного физико-химического процесса может совершаться работа. В данном случае это работа против силы тяжести. При достижении равновесия работа уже не совершается. Проведем мысленный эксперимент: добавляем в трубку осмометра воду. Столб жидкости становится выше равновесного, и далее наблюдается понижение уровня жидкости. Некоторое количество воды переходит через мембрану обратно в чистую воду. Это явление называется обратным осмосом.
Обратный осмос – это переход растворителя через мембрану из раствора в чистый растворитель под действием приложенного давления, превышающего осмотическое давление. Обратный осмос применяется для опреснения воды и концентрирования растворов.
Количественное изучение осмоса позволило установить закон осмотического давления, называемый законом Вант-Гоффа.
Осмотическое давление разбавленного раствора пропорционально его концентрации и температуре. Коэффициентом пропорциональности является универсальная газовая постоянная:
π = c·R·T
Принимая во внимание, что  , мы получаем выражение, совпадающее в уравнением состояния идеального газа:
, мы получаем выражение, совпадающее в уравнением состояния идеального газа:
 или
или 
Поэтому известна и такая формулировка закона Вант-Гоффа: осмотическое давление равно газовому давлению, которое производит газ при тех же параметрах состояния n, V и T. Осмотическое давление идеального раствора никак не зависит от его состава.
Применяя закон Вант-Гоффа, можно быстро оценивать осмотическое давление. Например, если в 1 л раствора содержится 1 моль вещества, то, учитывая аналогию с газом, напишем, или представим в уме выражение (по закону Бойля-Мариотта) 1 атм · 22,4 л = р атм · 1 л. Тогда π = р = 22,4 атм. Вообще, осмотическое давление оказывается, так сказать, большой силой, имеющей легко измеряемые значения даже для очень разбавленных растворов. Если бы в осмометре на рисунке находился раствор глюкозы с концентрацией 0,1 моль/л, то жидкость в трубке поднялась бы приблизительно на 25 м.
Осмотические измерения имеют также значение в медицинской диагностике и терапии. Одним из твердых правил инъекционных процедур является недопустимость введения в кровеносные сосуды дистиллированной воды. Вода смешивается с кровью, понижается осмотическое давление плазмы, и в эритроциты начинает поступать вода. Может начаться гемолиз. При патологических состояниях, сопровождающихся усиленным выделением воды, осмотическое давление плазмы крови может повышаться, что приводит к временной потере воды клетками. Это явление называется плазмолизом. Пределы изменения осмотического давления плазмы крови в норме составляют 730 – 780 кПа при физиологической температуре.
Пример. Имеется раствор глюкозы с массовой долей 0,5% (плотность 1,0001 г/л). Рассчитайте его осмотическое давление при 20ºС и температуру замерзания.
Решение. Для применения формул π = c·R·T и Δ Т з = K · b, следует вычислить молярную и моляльную концентрации, и найти в таблице криометрическую постоянную воды. Последняя равна 1,86 К·кг·моль–1. Для расчетов возьмем мысленно 1 л раствора; масса его составит 1,0001 кг. Очевидно, что при такой плотности, почти не отличающейся от плотности воды, молярная и моляльная концентрации будут различаться незначительно. Масса глюкозы в растворе
m (B) = 1000,1 г · 0,5%/100% = 5,0005 г
Количество вещества глюкозы (M = 180,155 г·моль–1) составляет
n = 5,0005 г/180,155 г·моль–1 = 0,02775665 моль
Теперь можно вычислить концентрации:
с = 0,02775665 моль/1 л = 0,02775665 моль/л
b = 0,02775665 моль·1000 г·кг–1/(1000,1 г – 5,0005 г) = 0,02789334 моль/кг
Уточненный расчет показал, что в данном случае разбавленного водного раствора, различие между молярной и моляльной концентрациями проявляется в третьей значащей цифре, причем оно обусловлено уменьшением содержания воды за счет присутствия растворенного вещества. Далее округлим концентрации до трех значащих цифр. Вычисляем осмотическое давление и температуру замерзания.
π = 0,0277 моль·л–1·8,31кПа·л·моль–1·К–1·(273,15 К + 20 К) = 67,5 кПа
Δ Т з = 1,86 К·кг·моль–1·0,0279 моль·кг–1 = 0,0519 К (также и ºС)
Т з = 0ºС – 0,0519ºС = –0,0519ºС
Результат расчета показывает, что осмотическое давление составляет 2/3 от атмосферы, и его можно измерить достаточно точно. Понижение температуры замерзания на 0,05 К измерить с такой же точностью значительно сложнее.
В практических исследованиях и измерениях приходится работать с растворами сложного состава (различные биологические жидкости). Такие растворы на основе осмометрии и криометрии (эбулиометрии) можно охарактеризовать суммарными значениями концентрации, которые называют осмолярность и осмоляльность.
На данном примере можно сказать, что если бы некий раствор имел π = 67,5 кПа и Δ Т з = 0,0519 К, то его осмолярность равнялась бы 0,0277 осмоль/л, и осмоляльность, соответственно 0,0279 осмоль/кг. При этом о качественном составе раствора нельзя бы сказать ничего определенного, так как π и Δ Т з – коллигативные свойства.
|
|
|
|
|
Дата добавления: 2013-12-13; Просмотров: 1006; Нарушение авторских прав?; Мы поможем в написании вашей работы!