КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Лекция 14 . Хроматографические методы анализа экотоксикантов
|
|
|
|
Хроматографические методы решают проблемы разделения сложной смеси на ее компоненты; определения идентичности и однородности химических соединений; количественного определения одного или нескольких компонентов сложной смеси; определения молекулярной структуры.
Газовая хроматография – наиболее широко используемый в анализе органических экотоксикантов метод аналитической химии. В основе метода лежат различия в распределении веществ между двумя фазами, из которых газовая является подвижной, а жидкая – неподвижной. В классической газовой хроматографии компоненты смеси переносятся подвижной фазой вдоль колонки, заполненной частицами твердого носителя, которые покрыты неподвижной фазой.
Элементы теории хроматографического процесса. Количественное описание процесса элюирования в газовой хроматографии наиболее просто может быть получено при кинетическом рассмотрении элементарных процессов движения молекул хроматографируемых соединений в колонке. Рассмотрим вкратце некоторые простые зависимости, определяющие скорость движения химических соединений через колонку, заполненную сорбентом с неподвижной жидкой фазой (вязкая органическая жидкость).
Каждое соединение имеет характерный для него коэффициент распределения (Г), определяемый уравнением:
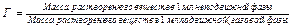 (1)
(1)
Коэффициент распределения Г определяет пропорцию, в которой анализируемые вещества (сорбат) распределяются между газом и жидкостью – неподвижной жидкой фазой (НЖФ). Как следует из формулы (1.1), коэффициент Г равен отношению концентрации анализируемого сорбата в жидкости (Сж) к концентрации его в газе (Сг). Таким образом, можно записать: Г = Сж/Сг. Следует помнить, что коэффициент Г – это постоянная величина для данного сорбата (вещества) в данной жидкости (неподвижной жидкой фазе), и она зависит только от температуры.
Продвигаясь в колонке в токе газа-носителя, анализируемые вещества сорбируются зернами сорбента (или растворяются в неподвижной фазе) и тут же «вымываются» из него этим же газом (десорбируются). Другими словами, хроматографический процесс есть цепь сорбционных и десорбционных актов, причем под сорбцией, как уже упоминалось выше, понимают либо растворение в неподвижной жидкости, либо адсорбцию на поверхности адсорбента, если он служит в колонке неподвижной фазой. Десорбция же – это обратимый процесс перехода вещества в газовую фазу. Газ при своем движении очищает колонку от сорбата, вымывает, элюирует его, или проявляет хроматограмму.
Для аналитика важно иметь такую колонку, которая как можно лучше делила бы компоненты анализируемой смеси. Разделение веществ в хроматографической колонке определяется двумя основными факторами: во-первых, селективностью неподвижной фазы, т.е. ее свойством по-разному сорбировать компоненты смеси, и, во-вторых, эффективностью колонки, т.е. способностью давать узкие хроматографические пики.
Задача исследователя, который желает разделить смесь на составляющие ее компоненты, заключается в том, чтобы подобрать такую НЖФ, которая по-разному растворяла бы эти компоненты. Другими словами, коэффициенты Г компонентов смеси для данной НЖФ должны быть разными, и чем больше они будут различаться, тем лучше – колонка будет хорошо делить анализируемые вещества.
По аналогии с ректификационной колонкой (например, используемой для перегонки нефти и получения бензина на нефтеперегонных заводах), эффективность хроматографической колонки оценивают числом теоретических тарелок (т.т.). В хроматографии эта величина определяется отношением времени удерживания вещества к ширине пика, т.е. скоростью размытия зоны. Чем меньше скорость размытия, тем более узкие пики на хроматограмме, тем эффективнее колонка. Эту зависимость можно представить в виде формулы:
n=5,545(tR /m0,5)2 (2)
где: п – число теоретических тарелок; tR – время удерживания; m0,5 – ширина пика на половине его высоты.
Сравнивая хроматографические колонки с ректификационными, мы видим, что последние по своей эффективности далеко позади. Самая обычная метровая колонка в хроматографии имеет эффективность в сотни т.т., в то время как эффективность заводской ректификационной колонны такой же длины на целый порядок меньше. Это и позволяет хроматографически разделять вещества, у которых значения Г различаются очень не намного, например, близкокипящие изомеры или молекулы, содержащие изотопы.
Эффективность колонки можно охарактеризовать и высотой, эквивалентной теоретической тарелке (ВЭТТ). Для известного числа т.т. ВЭТТ вычисляют по формуле:
Н = ВЭТТ = L/n (3)
где L – эффективная длина хроматографической колонки.
Если, как было показано выше, эффективность хроматографического разделения определяется относительным размыванием хроматографических зон в колонке и ее характеризуют числом теоретических тарелок п и высотой Н, эквивалентной теоретической тарелке (ВЭТТ), то степень разделения компонентов 1 и 2 характеризуетсякритерием разделения К:
Как видно из этой формулы, величина К равна разности времен удерживания, деленной на сумму ширин соседних пиков. Она, естественно, зависит от числа т.т. Величина К определяется селективностью сорбента и эффективностью хроматографической колонки. Чем длиннее колонка, тем выше ее эффективность. Зная значение входящих в эту формулу величин, можно определить, колонка какой длины нужна для разделения той или иной пары веществ. При К = 0 вещества не разделяются, но при К > 1 они полностью разделены, т.е. оптимальными являются условия, когда К близок к единице.
Если же, несмотря ни на что, вещества не разделяются, значит величины коэффициентов распределения у них одинаковы, и сколько не удлиняй колонку, эффекта не будет. Тогда нужно взять колонку с другой НЖФ, т.е. изменить селективность колонки. Тогда, как следует из уравнения 1.5, изменится величина К, так как изменится дробь в правой части этого уравнения, содержащая времена удерживания сорбатов (т.е. коэффициент селективности). Кстати, в ректификации (например, при перегонке бензинов) этого сделать нельзя, так как там степень разделения определяется лишь температурами разделяемых веществ.
Наличие эффективной колонки, позволяющей разделять многокомпонентные смеси, особенно важно в экологической аналитической химии, которая обычно имеет дело с очень сложными по составу композициями загрязняющих веществ различной природы.
Для перехода в газовую фазу образцы нагревают. В высокоэффективной, или капиллярной, газовой хроматографии применяются колонки без носителя, а тонкая пленка неподвижной фазы наносится на внутреннюю поверхность капилляра (WCOT-колонки). Это обеспечивает значительно большую эффективность разделения и меньший уровень фона по сравнению с насадочными колонками. Наибольшее распространение получили колонки из синтетического плавленого кварца, отличающегося высокой инертностью, необходимой для анализа пестицидов, органических кислот, аминов и др., а также гибкостью.
Выпускаемые в настоящее время промышленностью капиллярные колонки обычно имеют внутренний диаметр от 0,05 до 0,75 мм и длину от 30 до 105 м. Слой неподвижной фазы толщиной от 0,1 до 0,8 мкм наносят непосредственно на внутреннюю поверхность колонки или «пришивают» к ней химически. В качестве неподвижных фаз применяют полимеры, каучуки (OV-1, SE-30) или твердые вещества (карбовакс 20 М). Основные характеристики неподвижных фаз, используемых в капиллярных колонках, приведены в табл. 14.1.
Неподвижную фазу растворяют в соответствующем растворителе и наносят на внутреннюю поверхность капилляра динамическим или статическим методами, либо ковалентно связывают с поверхностью. Привитые фазы более долговечны и обладают большей термической устойчивостью по сравнению с исходными веществами. Кроме того, они не уносятся с потоком газа, что позволяет повысить верхний предел рабочих температур без заметного увеличения уровня фона. Колонки с привитыми фазами можно также промывать растворителями, тогда как колонки с нанесенными фазами промывать нельзя.
Таблица 14.1. Характеристики неподвижных фаз,
применяемых в капиллярной хроматографии
| Фаза | Состав | Полярность | Определяемые вещества | Диапазон температур |
| OV-1, OV-101, SP-2100, SE-30, DB-1, HP-1, SPB-1 | Полидиметил-силоксан | Неполярная | Фенолы, амины, пестициды, полихлорирован-ные бензолы, серосодержащие соединения | –60-320 |
| OV-73, SE-52, SE-54, SPB-5, HP-5, DB-5 | Поли(5% дифенил-, 95% диметил-)си- локсан | Тоже | ПХДД, ПХДФ, ПХБ, галогенсодержащие со-единения, пестициды | -60-32 |
| OV-7, SPB-20 | Поли (20% дифенил-, 80% диме-тил-) силоксан | Средняя | Ароматические углеводороды | -25-300 |
| OV-11,DB-35, SPB-35 | Поли(35% фенил-, 65% метил-)силок-сан | Тоже | Ароматические и полярные углеводороды, спирты | 0-300 |
| OV-1701, DB-1701, SPB-1701 | Поли(14% циано-пропилфенил-, 86% диметил-)силоксан | То же | Углеводы, спирты, пестициды, лекарственные вещества | До 280 |
| OV-2250, OV-17, HP-50, SPB-50,DB-17 | Поли(50%-дифе-нил-, 50%-диме-тил-)силоксан | То же | Лекарственные вещества, пестициды, гликоли | 30-310 |
| PAG, Pluronics F68 | Полиалкилен-гликоль | Полярная | Органические кислоты, простые эфиры, спирты | 30-220 |
| Nukol, HP-FFAP, DB-FFAP, SP-1000, OV-351 | Полиэтиленгли-коль, модифицированный нитротерефталевой кислотой | То же | Органические кислоты, лекарственные вещества | 60-200 |
| Supelkowax 10, Carbowax 20M, DB-WAX | Полиэтилен-гликоль | То же | Органические кислоты, метиловые эфиры, спирты | 50-280 |
| SP-2330, DB-23 | Поли(80%бис- цианопропил-, 20% цианопропил фенил-)силоксан | То же | Углеводы, эфиры | До 250 |
| SP-2340, OV-275 | Полибисциано-пропилсилоксан | То же | Углеводы, спирты, эфиры | 25-50 |
Основы метода. Схема простого газового хроматографа показана на рис. 14.1.
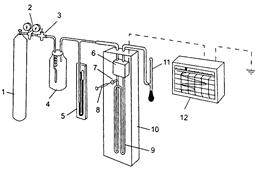
Рис.14.1. Простой газовый хроматограф
Газ-носитель из баллона (1) через редуктор (2), регулятор давления (3) и стабилизатор потока (4) поступает через сравнительную ячейку детектора 6 (если в качестве детектора используется катарометр) и затем через устройство для ввода пробы (7) в хроматографическую колонку (9), расположенную вместе с детектором в термостате (10).
Давление на входе в колонку измеряется манометром (5), объемная скорость газа-носителя периодически контролируется пенным измерителем скорости (11). Проба шприцем (8) вводится в поток газа-носителя перед хроматографической колонкой через устройство для ввода пробы (7). Поток газа-носителя переносит пробу в хроматографическую колонку (9), где и происходит разделение ее компонентов на отдельные зоны. Разделенные вещества (хроматографические зоны) поступают в детектор (б), который определяет концентрацию анализируемых компонентов в газе-носителе. Сигнал детектора, величина которого пропорциональна концентрации или потоку вещества, автоматически регистрируется потенциометром (12).
Основными узлами хроматографа являются хроматографическая колонка и детектор. Колонка выполняет функцию разделения анализируемой смеси на составные компоненты, а детектор количественно (в потоке газа-носителя) регистрирует концентрацию уже разделенных соединений. На рис. 3 схематично показаны отдельные этапы хроматографического разделения трехкомпонентной смеси («мгновенные фото» положений хроматографических зон в колонке через определенные интервалы времени) и связь процесса разделения с регистрируемой хроматограммой.
Хроматографические колонки. Хроматографические колонки, используемые для разделения смесей органических и неорганических веществ в газовой хроматографии, представлены в табл. 14.2
Таблица 14.2. Хроматографические колонки в газовой хроматографии
| Тип колонки | Материал | Область применения |
| Насадочные | Стекло, сталь | Газы, летучие соединения |
| Капиллярные WСОТ | Стекло, кварц | Летучие соединения |
| Капиллярные РLОТ | Кварц | Газы |
| Полнкапиллярные | Стекло | Летучие соединения (tкип. 40-400°С) |
Колонки заполнялись адсорбентом (адсорбционный вариант газовой хроматографии) – активным углем, силикагелем, оксидом алюминия и др. или сорбентом (газожидкостный вариант газовой хроматографии). Сорбент состоял из твердого диатомитового носителя (похожего на размолотый кирпич), на который наносилась в количестве 5-20% НЖФ (вязкая органическая жидкость). Эффективность таких колонок составляла несколько тысяч т.т., и они не позволяли добиться полного разделения многокомпонентных смесей органических соединений, какими являются смеси веществ, загрязняющих воздух, воду и почву.
В момент ввода анализируемой смеси зоны всех трех веществ расположены в начале хроматографической колонки. Под действием потока газа-носителя компоненты смеси начинают перемещаться вдоль колонки с различными скоростями, которые определяются природой разделяемых соединений и типом сорбента (рис. 14.2). Быстрее всех из колонки элюируется (выходит) компонент 1, окрашенный в черный цвет, затем компонент 2 (продольные линии) и последним компонент 3 (поперечные линии).
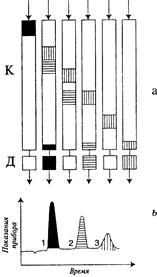
| Рис. 14.2. Схема хроматографического разделения для трехкомпонентной смеси: а – динамика хроматографическог процесса (положение хроматографических зон в колонке через определенные интервалы времени); б – регистрируемая хроматограмма. |
Разделение в газовой хроматографии основано на различном распределении молекул разделяемых компонентов между движущейся газовой фазой (газ-носитель) и неподвижной фазой (сорбент в колонке). Между этими фазами для каждого компонента анализируемой смеси в колонке устанавливается динамическое равновесие. Под действием потока газа-носителя компоненты анализируемой смеси с разными скоростями перемещаются вдоль хроматографической колонки. Скорость этого перемещения определяется для каждого компонента константой его распределения между газовой и неподвижной фазами. Скорость движения хроматографической зоны обратно пропорциональна константе распределения, т.е. хорошо сорбируемые компоненты передвигаются вдоль слоя сорбента медленнее, чем плохо сорбируемые. При продвижении смеси вдоль слоя сорбента сильно сорбирующиеся компоненты (с большой молекулярной массой, высокой температурой кипения) отстают от более легких (с небольшой молекулярной массой и более низкой температурой кипения) и появляются на выходе из колонки (где они фиксируются детектором) гораздо позднее легких компонентов. В результате компоненты смеси «растягиваются» во времени, образуя хроматограмму – график зависимости величины сигнала детектора от времени.
Рассмотрим основные элементы хроматограммы (рис. 14.3). Нулевая линия – участок хроматограммы (например, между пиками 1 и 2), получаемый при выходе из колонки только газа-носителя. Хроматографический пик – участок хроматограммы, соответствующий сигналу детектора во время выхода из колонки одного (или нескольких) компонентов.

| Рис. 14.3. Хроматограмма четырехкомпонентной смеси Условия разделения: хроматограф «Цвет-4», колонка 200х0,4 см, заполнена 10% апиезона К на хромосорбе Р, 65°С: 1 – воздух, 2 – циклогексадиен, 3 – циклогексан, 4 – метилциклогексан. |
Время удерживания (tR) – время, прошедшее от момента ввода пробы в колонку до выхода максимума пика.Ширина пика(m) – отрезок на нулевой линии, полученной интерполяцией нулевой линии в промежутке от начала до конца пика. В хроматографии чаще используют ширину пика на половине высоты (m0,5), практическое определение которой по хроматограмме проще.Высота пика (h) – расстояние от максимума пика до его основания, измеренное в направлении, параллельном оси сигнала детектора. Площадь пика (S) – площадь, заключенная между линией, ограничивающей пик, и его основанием.
Качественный анализ в газовой хроматографии проводится на основании измерения времен (или объемов) удерживания. Более удобно проводить идентификацию неизвестных компонентов, используя не абсолютные значения величин удерживания, а относительные величины. В частности,относительное время удерживания получают, деля время удерживания (время выхода из колонки) искомого компонента на время удерживания стандартного вещества. Относительное время удерживания мало зависит от изменения условий эксперимента (температура, давление газа-носителя и др.) и на его основе можно в некоторых случаях проводить идентификацию анализируемых соединений по каталогам времен удерживания.
Количественный анализ в газовой хроматографии основан на построение зависимости (гралуировочный график) площади пика (пиков) на хроматограмме (эта площадь прямо пропорциональна количеству вещества в хроматографической зоне от концентрации (С) компонента (компонентов) в анализируемой пробе.
Количественная интерпретация хроматограмм осуществляется методом внешнего стандарта. С этой целью ежедневно (в начале, в середине и в конце рабочего дня) снимают хроматограммы проб градуировочного раствора, который готовят ежемесячно и хранят в холодильнике. По результатам газохроматографического анализа проб, используя средние значения высот или площадей пиков, вычисляют количество анализируемого компонента в пробе.
Хроматографические детекторы. После хроматографической колонки разделенные компоненты поступают в детектор. Принципы действия детекторов могут быть самыми разными, но объединяет их одно – все они указывают на изменение какого-либо свойства газового потока в зависимости от состава анализируемой пробы. Сигнал детектора после усиления записывается на хроматограмме в виде пика, по которому судят о количестве вещества в пробе, а по временам удерживания (положение пиков на хроматограмме) иногда можно судить о качественном составе пробы.
В газовой хроматографии используется несколько десятков детекторов, из которых для экологических анализов (воздух, вода, почва) применяют около 10 детекторов (табл. 14.3).
Определение следовых количеств загрязняющих веществ в окружающей среде стало возможным лишь после появления пламенно-ионизационного детектора (ПИД). ПИД – универсальный чувствительный детектор, принцип действия которого основан на измерении электропроводности водородного пламени, которая резко возрастает при попадании в него ничтожных количеств органических веществ.
Таблица 14.3. Характеристики газохроматографических детекторов
| Обозначение | Тип | Селективность | Минимально детектируемое количество | Линейный диапазон |
| ПИД | Универсальный | — | 10 пг C/с | 107 |
| ЭЗД | Селективный | Вещества, содержащие атомы галогенов | 0,2 пг Cl/c | 104 |
| ТИД | Селективный | Азот- и фосфорсодержащие соединения | 1 пг N/c, 5 пг Р/с | 104 |
| ФИД | Селективный | Ароматические углеводороды | 107 | |
| ЭЛКД | Селективный | Соединения, содержащие атомы галогенов, S и N | 1 пг Cl/c, 5 пг S/c | 106 |
| МС | Универсальный | Характеристические ионы | 1 нг в режиме сканирования, | 104 |
| 1 пг в режиме масс-фрагментографии | 105 | |||
| АЭД | Универсальный | Любые вещества | 0,2-50 пг/с в зависи-мости от элемента | 104 |
| ИКС | Универсальный | Любые вещества, имеющие в ИК-спектре сильные полосы поглощения | 1 нг | 103 |
В принципе этот детектор напоминает обычную бытовую газовую горелку, только вместо газа в ней для горения используют водород. Газ-носитель (азот), поступающий в детектор из колонки, является прекрасным электроизолятором, но проводимость его существенно возрастает благодаря ионам, образующимся при горении органических соединений в водородном пламени. Элюат (в токе азота) смешивается в детекторе с водородом и поступает к соплу горелки, к которой одновременно подается очищенный воздух. Горение происходит между электродами, на которые подается напряжение порядка 100-300 В.
При сгорании органических веществ в пламени образуются заряженные частицы (ионы), а подаваемое на электроды напряжение заставляет ионы перемещаться от одного электрода к другому. Таким образом, возникает ионный ток, который измеряется электрометром и после усиления peгистрируется на хроматограмме в виде пиков.
Следует заметить, что во многом успех газохроматографического определения экотоксикантов зависит от применяемых детекторов. В частности, надежное газохроматографическое определение ПАУ на уровне следовых количеств возможно только с применением масс-селективного детектора, который представляет собой настольную модель квадрупольного масс-спектрометра. В табл. 14.4 приведены основные характеристики детекторов, используемых в газовой хроматографии. Наиболее важными из них являются минимально детектируемое количество, диапазон чувствительности и селективность.
Таблица 14.4. Характеристики детекторов,
используемых в газовой хроматографии
| Детектор | Определяемые соединения | Минимальное детектируемое количество, пг | Линейный диапазон |
| Пламенно-иониза-ционный детектор (ПИД) | Углеводороды | 107 | |
| Детектор электронного захвата (ДЭЗ) | Галогенсодержащие органические соединения | 0,2 | 104 |
| Термоионный детектор (ТИД) | Азот- и фосфорсодержащие органические соединения | 1 (N); 5 (Р) | 104 |
| Фотоионизационный детектор (ФИД) | Ароматические углеводороды | 107 | |
| Масс-селективный детектор (МСД) | Характеристические ионы | 105 | |
| Атомно-эмиссион-ный детектор (АЭД) | Любые вещества | 0,2-50 | 104 |
| ИКС-детектор | Тоже | 103 |
Наиболее универсальным является пламенно-ионизационный детектор (ПИД), работа которого основана на измерении ионного тока, протекающего между электродами при попадании в водородное пламя органических соединений. Однако селективность ПИД недостаточна для анализа сложных препаратов.
Для определения галогенсодержащих соединений, таких как ХОП, ПХБ, ПХДД и ПХДФ, в основном используется детектор электронного захвата (ДЭЗ), принцип действия которого основан на уменьшении проводимости, вызываемом захватом электронов определяемым веществом. В состав детектора входит радиоактивный источник (обычно 63Ni), который испускает электроны высокой энергии. Наряду с ПИД и ДЭЗ для определения летучих ароматических и галогенсодержащих соединений в последнее время рекомендуют использовать фотоионизационный детектор (ФИД). При прохождении через ФИД молекулы органических соединений возбуждаются УФ-лампой с образованием заряженных частиц, которые формируют электрический ток.
Термоионный детектор (ТИД) является модификацией ПИД, в котором для селективной ионизации в водородном пламени органических соединений, содержащих атомы азота и фосфора, используется таблетка или шарик из рубидиевого стекла. ТИД широко применяется при определении гербицидов, инсектицидов и фунгицидов. Однако более удобен для этих целей атомно-эмиссионный детектор (АЭД) с микроволновой плазмой. В АЭД выходящие из колонки вещества атомизируются и возбужденные атомы излучают свет, интенсивность которого измеряется в фотодиодной матрице. Поскольку каждый химический элемент имеет свой спектр эмиссии, то обеспечивается исключительно высокая селективность аналитического сигнала.
В случае ИКС-детекторов последовательно регистрируются ИК-спектры элюируемых из колонки соединений. Поток газа-носителя поступает в кювету, в которой молекулы поглощают ИК-излучение с точно определенной частотой. Чувствительность детектирования зависит от наличия в органических соединениях тех или иных функциональных групп. Если молекула хорошо поглощает ИК-излучение, то аналитический сигнал может быть получен при поступлении в кювету 1 нг вещества. Следует заметить, что комбинация ГХ с ИКС и МС является в настоящее время самым мощным инструментом для идентификации экотоксикантов. Это связано с тем, что спектр ИК-поглощения очень специфичен, и служит основой для определения вещества.
Современные газовые хроматографы представляют собой многодетекторные полностью автоматизированные приборы, в которых все стадии анализа регулируются компьютером. Методом газовой хроматографии анализируют газы и летучие (органические и неорганические) соединения, которые можно перевести в газовую фазу. Высококипящие (высокомолекулярные) соединения, трудно превращаемые в пар (газ), исследуют методом жидкостной хроматографии.
Ионная хроматография. Ионная хроматография (ИХ) является высокоэффективным вариантом ионообменной хроматографии (ИОХ). Ионный обмен – обратимый процесс стехиометрического обмена между двумя контактирующими фазами – раствором электролита (соли) и ионитом (сорбентом).
Большое значение ионный обмен имеет в агрохимии, процессах жизнедеятельности и химическом анализе. Метод ионообменной сорбции применяют для умягчения или обессоливания воды (например, для опреснения морской воды), удаления солей из сахарных сиропов, молока, вин, растворов фруктозы, дубильных веществ, продуктов гидролиза сельскохозяйственного сырья, растворов лекарственных препаратов (антибиотиков, витаминов, алкалоидов), для удаления ионов кальция из плазмы крови перед ее консервацией, для очистки от минеральных ионов растворов органических реагентов, для очистки сточных вод от фенола и тяжелых металлов, а также для извлечения (концентрирования) пенных ионов, находящихся в микродозах в растворе (например, редкоземельных элементов). Ионный обмен широко применяют в гидрометаллургии – для извлечения благородных, цветных и редких металлов из сточных вод (например, ионов из стоков гальванических цехов), для улавливания и концентрирования радиоактивных ионов и ионов меди из стоков медноаммиачного производства искусственного шелка
Механизм разделения в ионообменной хроматографии. Одной из наиболее важных областей применения ионного обмена является ионообменная хроматография – разделение сложной смеси электролитов в разбавленном растворе. Хроматографическую колонку заполняют ионитами – ионообменными сорбентами минерального происхождения (силикаты, алюмосиликаты) или синтетическими полимерными органическими сорбентами (полистирольными, фенолформальдегидными катионитами или аминоформальдегидными и полиаминовыми анионитами). Наиболее распространенным является взгляд на механизм ионного обмена как гетерогенную химическую реакцию двойного обмена:
2RH + CaCl2 «RH + Ca2+ + 2Cl– «R2Ca + HCl
ионит электролит
Как видно из этого уравнения, неорганические или органические ионы поглощаются ионитом, но если потом промыть колонку раствором кислоты (правая часть уравнения), реакция пойдет в другую сторону (справа налево). В результате удерживаемые ионитом ионы снова перейдут в раствор. Этот процесс сорбции и последующего вытеснения ионов и является собственно хроматографическим разделением в ИОХ. Иными словами, процесс хроматографирования в ИОХ состоит в том. что в колонку вводят небольшое количество анализируемого раствора (смесей ионов) и промывают ее специально подобранным растворителем. Разделенные ионы перемешаются вдоль хрома-тографической колонки с различными скоростями в зависимости от их сродства к иониту. и на выходе из колонки детектируются, например, с помощью УФ-детектора.
Ионообменную хроматографию широко применяют при разделении близких по свойствам ионов – смесей РЗЭ, гафния. ниобия, циркония, таллия и др., изотопов, антибиотиков, для получения чистых органических препаратов (карбоновых кислот, аминокислот, алкалоидов, витаминов, антибиотиков и др.).
Разделение различных смесей неорганических ионов с помошью ИОХ давно уже стало традиционным методом в практической аналитике. Тем не менее подобные методики до последнего времени имели ограниченное применение. Это объясняется невысокой скоростью элюирования (длительный анализ) и значительным размыванием хроматографнческих зон (широкие пики на хроматограммах) на ионообменных сорбентах. Трудности были связаны и с детектированием ряда ионов. Все это-не позволяло быстро и эффективно проводить анализ. В ИХ происходит ионообменное разделение на первой (разделительной) колонке с последующим подавлением фонового сигнала на второй (подавляющей) колонке. Ионный хроматограф состоит из резервуара с элюентом, насоса высокого давления, разделяющей и подавляющей колонок, детектора (чаще всего кондуктометрического) и регистрирующего устройства.
Разделение ионов методом ИХ можно осуществлять и на жидкостных хроматографах, но в настоящее время ряд фирм (в том числе и в России) выпускают специальные ионные хроматографы. Их широко используют в геологии, энергетике, пищевой и фармацевтической промышленности, медицине и анализе объектов окружающей среды. Последнее обусловлено особенностями и достоинствами ИХ:
– возможность одновременного определения большого числа неорганических и органических ионов, а также одновременного определения катионов и анионов;
– низкий предел обнаружения (до 1 нг/мл без предварительного концентрирования);
– высокая селективность определения ионов в сложных смесях и экспрессность (можно определять 10 ионов за 15-20 мин.);
– широкий диапазон определяемых содержаний (от 1 нг/мл до 1000 мг/л без разбавления);
– малый объем анализируемой пробы (0,1-0,5 мл);
– простота подготовки пробы к анализу (часто возможен и прямой анализ, без пробоподготовки);
– возможность использования различных детекторов и их комбинаций, что позволяет обеспечить высокую селективность определения и надежные результаты идентификации целевых компонентов.
Одно из важнейших направлений использования ИХ в экологической аналитической химии — анализ вод. Известно, насколько важно обнаружить вредные примеси в питьевой и водопроводной воде, в атмосферных осадках, речной и морской роде, подземной и поверхностной водах, а также установить степень загрязненности сточных и сбросовых вод. Среди этих компонентов существенное место занимают неорганические ионы, ионы металлов, ионогенные органические соединения.
Варианты ионной хроматографии. Рассмотрим некоторые типичные варианты анализа ионов, которые применяются в практической аналитике и при решении задач экологической аналитической химии.
Двухколоночная схема с кондуктометрическим детектирование. Двухколоночная ИХ является классическим вариантом этого метода. Этот способ детектирования ионов (рис. 14.4) в принципе обеспечивает наибольшую чувствительность. Однако практическое достижение высокой чувствительности затруднено высокой электропроводностью типичных подвижных фаз (элюентов), используемых в ИОХ (что мешает работе кондуктометрического детектора — детектора по электропроводности).
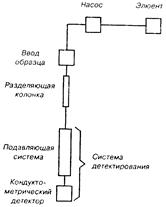 Рис. 14.4. Схема двухколоночной ионохроматографической системы.
Рис. 14.4. Схема двухколоночной ионохроматографической системы.
|
 Рис. 14.5. Схема одноколоночной ионохроматографической ситсемы.
Рис. 14.5. Схема одноколоночной ионохроматографической ситсемы.
|
Для снижения электропроводности между разделительной к донкой и кондуктометрическим детектором (КД) устанавливают вспомогательную (подавляющую) ионообменную колонку, нейтрализующую элюат и снимающую (вычитающую) его электропроводность. На фоне обработанной таким образом подвижной фазы достигается высокая чувствительность анализа. Переключение потоков, необходимое для периодической регенерации подавляющей колонки, осуществляется с помощью автоматизированных устройств, входящих в состав серийных ионных хроматографов.
Одноколоночная схема (рис. 14.5) уступает по чувствительности рассмотренной выше. Ее преимуществом является возможность работы на обычном оборудовании для ВЭЖХ, имеющемся во многих лабораториях. Помимо простоты аппаратуры (не нужна подавляющая колонка при использовании элюентов малой проводимости) для одноколоночной схемы характерна более высокая эффективность разделения, более широкий выбор сорбентов (например, на основе силикагеля) и элюентов, а также возможность использования различных детекторов, которые применяются в ВЭЖХ.
Типичные хроматограммы разделения неорганических катионов и анионов представлены на рис. 14.6-14.8. Разделение галоген-, нитрат-, нитрит-, фосфат- и сульфат-ионов (рис. 14.6) осуществляли на полимерном макропористом геле. Это позволяет достичь высокой эффективности разделения (20000 тт/м) и в течение 7 мин разделить 7 неорганических анионов.
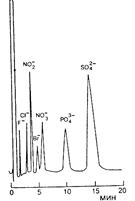
| Рис. 14.6 Разделение неорганических анионов на анионообменном сорбенте TSK GEL 620. Элюент 1,3 мМ Na2B4О7/5,8 мМ Н3ВО3/1,4 мМ глюконат калия (рН 8,5) в H2O – СН3СН (88:12). Скорость потока 1,2 мл/мин. Концентрация анионов 5-40 мкг/мл. Детектирование кондуктометрическое. |
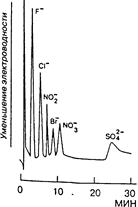 Рис.14.7. Хроматограмма неорганических анионов. Колонка TSK GEI IC – Anion-PW. Элюент 2 мМ КОН. Скорость потока 1,0 мл/мин. Концентрация анионов 10 мкг/мл. Детектирование косвенное кондуктометрическое.
Рис.14.7. Хроматограмма неорганических анионов. Колонка TSK GEI IC – Anion-PW. Элюент 2 мМ КОН. Скорость потока 1,0 мл/мин. Концентрация анионов 10 мкг/мл. Детектирование косвенное кондуктометрическое.
| 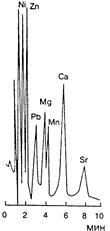 Рис.14.8 Хроматограмма двузарядных катионов. Колонка Dionex CS2 4х250 мм. Элюент 2 мМ лимонная кислота/2 мМ этилендиамин (рН 4,0). Скорость потока 1,5 мл/мин. Концентрация катионов 5-20 мкг/мл. Детектирование кондуктометрическое.
Рис.14.8 Хроматограмма двузарядных катионов. Колонка Dionex CS2 4х250 мм. Элюент 2 мМ лимонная кислота/2 мМ этилендиамин (рН 4,0). Скорость потока 1,5 мл/мин. Концентрация катионов 5-20 мкг/мл. Детектирование кондуктометрическое.
|
Для разделения катионов используют поверхностно-сульфированные катионообменники на основе сополимера стирол-дивинилбензол. Более всего в одноколоночной ИХ распространены сорбенты на основе силикагеля. На них можно разделять как катионы. так и анионы, используя в качестве элюентов растворы фталевой, бензойной и салициловой кислот и их смеси.
Всего 10 мин требуется для разделения 7 катионов (рис. 14.8), а общее число катионов и анионов органической и неорганической природы, которые можно определять методом ИХ, составляет несколько десятков. По этой причине метод ИХ является альтернативным для газовой хроматографии, в которой для определения анионов их предварительно переводят в летучие органические производные.
Пока одноколоночный вариант ИХ используют реже двухколоночного. Тем не менее одноколоночное определение неорганических ионов в природных и промышленных объектах (вода, почва, донные отложения и др.) характеризуется высокой селективностью, экспрессностью и воспроизводимостью (Sr не превышает 6%). Пробоподготовка в большинстве случаев заключается лишь в фильтровании образца воды через пористый фильтр с размером пор 0,45 мкм. Предел обнаружения (определения) анионов 100-300 мкг/л, что в 10-50 раз больше предела определения большинства анионов двухколоночным методом.
Детекторы в ионной хроматографии. Чаще всего в ИХ применяют кондуктометрический детектор, схема которого изображена на рис. 14.9. Если к двум электродам, находящимся в растворе электролита, приложено напряжение, то в растворе возникает электрический ток, а проводящая среда имеет электрическое сопротивление. На измерении этого сопротивления основан принцип действия большинства кондуктометрических детекторов.
Детектор состоит из проточной ячейки, в которую подается анализируемый раствор, индикатора и системы регистрации кондуктометрического сигнала. Кондуктометрическая ячейка представляет собой камеру объемом менее 10 мкл, соединенную с двумя электродами (3 и 5) из платины, золота или нержавеющей стали. Сопротивление ячейки, как правило, измеряют с помощью моста сопротивлений Уитстона.
Помимо КД в ИХ используют многие детекторы, применяемые в ВЭЖХ. Это, в частности, рефрактометр, ионселективные электроды, атомно-абсорбционный спектрометр (ААС), масс-спектрометрический и электрокинетический детекторы.
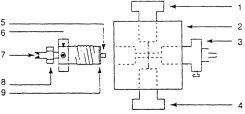
Рис. 14.9. Схема кондуктометрической ячейки фирмы Biotronik. 1 – вход элюента; 2 – корпус ячейки; 3 – электрсл; 4 – выход элюента; 5 – позолоченный электрод;
6 – контакт с измерительным устройством; 7 – болт для изменения постоянной ячейки; 8 – контрогайка; 9 – уплотнительное кольцо.

Рис.14.10. Хроматограммы анионов, полученные на колонке Ионосфер А (250 х 4,6 мм), подвижная фаза – 0,04 М бифталат калия, рН 4,0, детекторы – рефрактометр (а) и кондуктометр (б).
Для надежной идентификации разделенных ионов помимо характеристик удерживания важен тип применяемого детектора, особенно его селективность по отношению к целевым компонентам. Лучшие результаты в смысле их надежности могут быть получены при использовании сочетаний детекторов, например, рефрактометра и КД.
Надежная идентификация может быть реализована и с помощью селективных детекторов. Так, для детектирования фторид- и бромид-ионов лучшим является детектор на основе ионселективных электродов. С помощью ААС определяли низкие содержания органических и неорганических соединений мышьяка, причем предел обнаружения в пробе воды 0,5 л составлял 0,01-0,001 мг/л.
Количественное определение ионов методом ИХ проводят точно так же, как и в случае ВЭЖХ.
Хромато-масс-спектрометрия. Основной принцип хромато-масс-спектрометрии состоит в хромато-графическом разделении определяемых соединений, их ионизации и детектировании ионов по величине отношения массы к заряду, которое осуществляется в масс-спектрометре. Соединение хроматографа с масс-спектрометром – это не просто объединение двух разных приборов для решения одной задачи, а появление нового метода со своими особенностями и возможностями. Его можно использовать даже в тех случаях, когда вещества не удается разделить хроматографически. Как правило, в хромато-масс-спектрометрах используются серийные газовые хроматографы с капиллярными колонками (ГХ-МС). При этом разделение смеси веществ осуществляется методом газовой хроматографии, а масс-спектрометр выполняет роль высокоэффективного детектора. Очевидно, что хромато-масс-спектрометрия в варианте ГХ-МС применима для определения только тех веществ, которые имеют достаточно высокую летучесть и термически стабильны. Последнее обстоятельство является немаловажным ограничением, что стимулировало разработку приборов, в которых масс-спектрометр сочетают с жидкостным хроматографом (ЖХ-МС).
После хроматографического разделения молекулы образца ионизируются в вакууме или в атмосфере инертного газа. В настоящее время чаще всего используют ионные источники, в которых определяемое вещество ионизируется под действием пучка электронов, испускаемых раскаленным рениевым или вольфрамовым нитевидным катодом и ускоряющихся в электрическом поле (электронный удар). Для предотвращения конденсации вещества на стенках ионизационной камеры ее обычно нагревают до 200-250°С. При соударении электронов с молекулами образца последние ионизируются.
Для большинства органических соединений потенциалы ионизации находятся в диапазоне от 7 до 20 эВ, поэтому стандартные ионные источники генерируют электроны с энергией около 70 эВ, что обеспечивает ионизацию любых молекул. Образующиеся молекулярные ионы имеют относительно большую внутреннюю энергию и распадаются, причем характер фрагментации зависит от величины этой энергии, структуры исходных соединений, локализации заряда и наличия «слабых» мест в структуре. Фрагментация может протекать настолько быстро, что масс-спектры некоторых соединений не содержат пиков молекулярных ионов. Это один из наиболее существенных недостатков ионизации электронным ударом. Однако для масс-спектров, полученных ионизацией электронным ударом, имеются большие базы данных в виде каталогов и атласов (даже электронные библиотеки).
В случае химической ионизации сначала ионизируется газ-реагент (Аr, NН3, Н2, CH4, изо- С4H10, О2, H2O и др.), находящийся под давлением 0,2-2 мм рт. ст., а затем возникающие ионы, например СH5–, реагируют с молекулами определяемого соединения, в результате чего образуются ионы типа MH+ или М+, M2+ и др. Ионно-молекулярные реакции протекают в более мягких условиях, а образующиеся молекулярные ионы более устойчивы. По этой причине масс-спектры при химической ионизации проще, чем при электронном ударе.
Существуют три типа основных процессов, приводящих к образованию отрицательных ионов: 1)ионно-молекулярные реакции между нейтральными молекулами и анионами: М + X– ® MX–; 2) резонансный захват электронов (тепловых) с образованием молекулярных анионов: М + е– ® М–; 3) диссоциативный захват электронов с образованием осколочного аниона и нейтральной частицы: АВ + е– ® А– + В.
Масс-спектры, получаемые при отрицательной химической ионизации, более просты, чем при ионизации электронным ударом. Кроме того. образование отрицательных ионов позволяет повысить чувствительность детектирования соединений с высоким сродством к электрону по сравнению с масс-спектрометрией положительных ионов в 10-100 раз, причем линейная зависимость величины сигнала от количества вещества сохраняется в диапазоне, верхняя граница которого на 3 порядка превышает нижнюю границу. В частности, для хлорированных углеводородов предел обнаружения достигает»10 фг. В результате пределы обнаружения ПХДД и ПХДФ удалось снизить до уровней, приведенных в табл. 14.5.
Таблица 14.5. Пределы обнаружения диоксинов в различных объектах
| Объект | Единица измерения | Предел обнаружения |
| Воздух | пг/м3 | 0,0001 |
| Зола | нг/кг | 0,1-1,0 |
| Почва | Тоже | 0,1-1,0 |
| Донные отложения | Тоже | 0,1 |
| Вода | пг/л | 0,01-0,5 |
| Молоко | нг/л | 0,1-0,5 |
| Рыба | нг/кг | 0,1-1,0 |
| Жировая ткань | То же | 2-10 |
| Плазма крови | Тоже | 0,02-0,003 |
| Печень | Тоже | 1,0 |
Для количественного изомер-специфического определения ПХДД и ПХДФ на фоновом уровне обычно используют магнитные масс-спектрометры высокого разрешения. В этих приборах ионы, движущиеся в постоянном магнитном поле, отклоняются в направлении, перпендикулярном направлению движения, и меняют траекторию в зависимости от скорости. Последняя в свою очередь определяется энергией ионов и отношением массы к заряду. Одновременно магнитное поле фокусирует ионные пучки, отличающиеся угловым распределением. Благодаря данному обстоятельству ионы с различной величиной отношения т/z могут быть сфокусированы поочередно на входной щели детектора путем изменения величины магнитного поля.
Для детектирования ионов применяют электронные умножители с большим коэффициентом усиления, быстродействием и сравнительно малым шумом. Их недостатком является «старение» со временем и в результате загрязнения. В последнее время все большее распространение получают способы регистрации с помощью матричных и электронно-оптических систем. Первые представляют собой плоскостные системы из 104-107 электронных умножителей диаметром 10-12 мкм с расстоянием между соседними элементами ~15 мкм, а вторые – пластины, на которых падающие ионы с помощью фосфоресцирующего экрана создают видимое изображение масс-спектра. Такие системы позволяют одновременно регистрировать до 4% диапазона масс и обеспечивают разрешение до 5000. Для точного измерения масс ионов приборы градуируют с помощью стандартов.
Применение масс-спектрометрии в сочетании с хроматографией дает дополнительные возможности при определении органических экотоксикантов в объектах окружающей среды. Благодаря тому, что масс-спектрометр является высокоселективным детектором, разрешение пиков на масс-хроматограммах, как правило, заметно лучше, чем на обычных хроматограммах. Кроме того, по масс-хроматограммам можно получить ответ о природе анализируемых соединений. Это необходимо при идентификации загрязнителей, присутствующих в ультрамалых количествах.
Количество исследуемого компонента определяется по площади его пика на масс-хроматограмме, величина которой пропорциональна концентрации. В качестве аналитического сигнала иногда используют не один, а несколько пиков (для улучшения отношения сигнал/шум). Для построения градуировочных графиков применяют внешние или внутренние стандарты. Последние обеспечивают более точные результаты, поскольку в этом случае аналитические данные не зависят от изменения условий анализа. В качестве внутренних стандартов применяют индивидуальные соединения, близкие по характеристикам (время удерживания, летучесть, молекулярная масса и др.) к определяемым веществам, но отличающиеся от них по положению аналитического сигнала на масс-хроматограммах. Очень часто для этих целей применяют фторированные аналоги исследуемых соединений. Наиболее подходящими стандартами, ближе всего соответствующими анализируемым веществам, являются изотопно-меченые соединения (13С, 2H). При этом они должны удовлетворять следующим условиям: 1) пики внутренних стандартов должны быть в соответствующих местах на всех хроматограммах; 2) не должно быть наложений на пики внутренних стандартов; 3) соотношение пиков изотопов должно укладываться в необходимые пределы; 4) отношение сигнал/шум – не менее 10:1; 5) эффективность извлечения внутренних стандартов из препарата – от 40 до 150%.
Высокоэффективная жидкостная хроматорафия. Метод ВЭЖХ в последние годы по праву считается одним из наиболее важных в аналитической химии для определения следовых количеств пестицидов и ПАУ, в анализе пищевых продуктов и фармацевтических препаратов. В отличие от ГХ этот метод используется для определения термически нестойких, нелетучих или очень полярных соединений, но в то же время может быть применен и для анализа веществ, которые обычно определяют с помощью высокоэффективной газовой хроматографии.
Для эффективного хроматографического разделения определяемых компонентов наиболее часто применяют колонки длиной 25 см и внутренним диаметром 4-5 мм, заполненные сферическими частицами силикагеля размером от 5 до 10 мкм с привитыми октадецильными группами. Появление в последние годы колонок меньшего диаметра, заполненных более мелкими частицами силикагеля, привело к уменьшению расхода растворителей и продолжительности анализа, увеличению эффективности разделения.
Большое значение в ВЭЖХ имеет выбор стационарной фазы. В общем случае прочность удерживания разделяемых компонентов зависит от энергии адсорбции молекул растворителя и растворенных веществ. Чем больше энергия адсорбции последних, тем прочнее удерживаются разделяемые компоненты в колонке. Соответствующие значения энергий адсорбции зависят от вида взаимодействия, определяемого природой поверхности и адсорбирующихся веществ. Наиболее распространенные полярные сорбенты - силикагели, целлюлоза, оксид алюминия. Они применяются для определения афлатоксинов, лекарственных веществ, нитрозаминов. Заметим, что присутствие воды оказывает существенное влияние на свойства полярных сорбентов из-за ее конкурентной сорбции. Поэтому содержание воды в таких сорбентах поддерживают постоянным, не допуская длительного контакта с атмосферным воздухом.
Среди обращенно-фазовых сорбентов на основе силикагелей максимальный эффект обращения полярности достигается при прививке алкильных групп. При этом свойства обращенно-фазовых сорбентов зависят не только от природы привитых групп и удельной поверхности, но и от структуры привитого слоя. По этому признаку они делятся на три основных типа:
– с мономолекулярным слоем привитых функциональных групп;
– с поверхностным полимерным слоем;
– с объемно-модифицированным слоем,
В ВЭЖХ наибольшее практическое применение получили сорбенты первою типа с «щеточными» структурами привитых алкильных групп, содержащими от 1 до 22 метильных звеньев. При большой длине цепи алкильные группы изгибаются, заполняя неровности поверхности. Такие фазы приближаются по свойствам к жидким с тем преимуществом, что они не уносятся потоком растворителя. Модифицирование дифенильными группами повышает селективность неподвижной фазы по отношении к ароматическим соединениям. Прочность удерживания разделяемых компонентов возрастает с увеличением длины алкильных радикалов. Для определения неполярных молекул большой массы лучшие результаты обеспечивают фазы с короткоцепными алкильными группами.
Выбор подвижной фазы, как правило, основывается на эмпирическом подборе индивидуальных растворителей или их смесей, имеющих необходимую элюирующую способность. Последнюю выражают способностью растворителя взаимодействовать с адсорбентом. В случае полярных фаз относительная активность растворителей, как правило, сохраняется при переходе от одного адсорбента к другому, что позволяет расположить их в элюотропный ряд (табл. 14.6). Для сорбентов с обращенной фазой эта последовательность обратная.
Таблица 14.6. Элюирующая способность растворителей (e0)
для оксида алюминия
| Растворитель | e0 | Растворитель | e0 |
| н -Пентан | 0,00 | Метилэтилкетон | 0,51 |
| Изооктан | 0,01 | Ацетон | 0,56 |
| н -Декан | 0,04 | Этилацетат | 0,58 |
| Пентен | 0,08 | Анилин | 0,62 |
| Четыреххлористый углерод | 0,18 | Диэтиламин | 0,63 |
| Ксилол | 0,26 | Ацетонитрил | 0,65 |
| Изопропиловый эфир | 0,28 | Пиридин | 0,71 |
| Толуол | 0,29 | н -Пропанол | 0,82 |
| Бензол | 0,32 | Изопропанол | 0,82 |
| Этиловый эфир | 0,38 | Этанол | 0,88 |
| Хлороформ | 0,40 | Метанол | 0,95 |
| Тетрагидрофуран | 0,45 | Этиленгликоль | 1,11 |
Для обнаружения анализируемых компонентов в ВЭЖХ широко применяются устройства, работа которых основана на измерении поглощения в ультрафиолетовой области, флуоресценции или электрохимических характеристик. Возможно также сочетание жидкостного хроматографа с масс-спектрометром.
Детекторы для ВЭЖХ. В отличие от газовой хроматографии, в жидкостной хроматографии применяют гораздо меньше детекторов (не более 20 типов), которые можно разделить на оптические, электрические и электрохимические.
Универсальным детектором для ВЭЖХ является рефрактометрический детектор (РМД), принцип действия которого основан на дифференциальном измерении показателя преломления чистого растворителя и раствора анализируемого вещества в этом растворителе. Однако недостаточно высокая чувствительность и несовместимость с градиентами давления делают его значительно менее популярным в сравнении с ультрафиолетовым детектором.
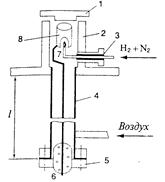
| Рис. 14.11. ПИД с выносным изолятором. 1 – крышка; 2 – корпус; 3 – изолятор; 4 – термостат; 5 – фланец; 6 – выносной изолятор; 7 – горелка; 8 – анод; l – длина выносного изолятора (около 2,00 мм). |
Ультрафиолетовый детектор (УФД) относится к селективным детекторам, так как реагирует только на вещества, поглощающие свет в УФ-области спектра (190-380 нм). В настоящее время более 60 фирм серийно производят ультрафиолетовый абсорбционный детектор с фиксированной или переменной длиной волны. Этот детектор гораздо чувствительнее рефрактометрического детектора, но высокочувствительная запись спектров стала реальностью лишь недавно — с началом использования детектора на диодной матрице (ДМД), работающего как в ультрафиолетовой, так и в видимой области спектра (так называемый УФ-В-детектор). В таком детекторе (рис. 14.12) «матрица» фотодиодов (их более 200) постоянно регистрирует сигналы в УФ- и видимой части спектра, обеспечивая таким образом запись УФ-В-спектров в режиме сканирования.
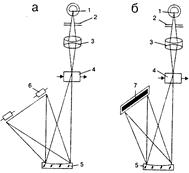
| Рис. 14.12. Фотодиодные УФ-детекторы для жидкостной хроматографии с механическим движущимся фотодиодом (а) и с фотодиодной матрицей (б): 1 – источник УФ-излучения; 2 – диафрагма; 3 – конденсор; 4 – проточная ячейка; 5 – дифракционная решетка; 6 – фотодиод; 7 – фотодиодная матрица. |
Это позволяет непрерывно снимать при высокой чувствительности неискаженные спектры быстро проходящих через спектральную ячейку компонентов.
Данные, полученные от ДМД одновременно на различных длинах волн, обрабатываются и оцениваются с помощью программного обеспечения, которое производит поиск в спектральной библиотеке, выделяет сигнал на определенной длине волны для повышения селективности, вычитает фон и осуществляет другие операции. Программное обеспечение может быть использовано для автоматической сверки УФ-спектров с известными, для идентификации компонентов и проверки «чистоты» пиков путем сравнения спектров, записанных при начале и окончании выхода каждого пика.
Такая автоматизированная система слежения идеальна для скрининга, поскольку позволяет аналитику отобрать для дальнейших исследований только «подозрительные» пики. В специально сконструированных для колонок малого объема УФ-В-детекторах на диодной матрице используются проточные ячейки, позволяющие производить запись полных УФ-В-спектров на диапазонах чувствительности, которые еще совсем недавно были трудно достижимы даже на детекторах с фиксированной длиной волны. Это существенное снижение пределов детектирования прямо повлияло на качество результата, позволяя аналитику проверять, является ли вещество, концентрацию которого он определяет, искомым компонентом. По сравнению с детектированием на одной длине волны, которое не дает информации о «чистоте» пика, возможности сравнения полных спектров диодной матрицы обеспечивают получение результата идентификации с гораздо большей степенью достоверности.
Ультрафиолетовый детектор на диодной матрице имеет низкий предел детектирования. В сочетании с извлечением токсичных примесей из воды методом твердофазной экстракции могут быть достигнуты относительно высокие уровни обогащения пробы. Например, определение пестицидов становится возможным при их содержании в питьевой воде на уровне 0,05 мкг/л и ниже. Этот детектор незаменим при определении очень низких содержаний/пестицидов, гербицидов, фенолов, ПАУ и других приоритетных загрязнителей воды, воздуха и почвы.
Флуоресцентный детектор (ФЛД) является вторым по популярности детектором в жидкостной хроматографии после ультрафиолетового; более 40 фирм оснащают ФЛД выпускаемые ими жидкостные хроматографы. Принцип действия ФЛД основан на измерении не поглощенного (как в УФ-детекторе), а испускаемого молекулами света (электромагнитного излучения). Молекулы некоторых соединений излучают часть поглощенной радиации (обычно в видимом диапазоне) в форме излучения более низкой энергии, с большей длиной волны. Это излучение может быть измерено и использовано для определения концентрации вещества.
Большая популярность ФЛД объясняется очень высокой селективностью и чувствительностью, и тем фактом, что многие загрязнители окружающей среды (например, ПАУ) флуоресцируют. Длины волн возбуждения и испускания могут быть выбраны независимо, и сам флуоресцентный сигнал обеспечивает гораздо более высокую чувствительность по сравнению с УФ-В-детектором (в последнем случае сигнал детектора представляет собой разницу между полной интенсивностью света источника и света, ослабленного за счет поглощения молекулами вещества, проходящего через ячейку детектора).
В флуоресцентном детекторе сигнал не возникает до тех пор пока способное флуоресцировать вещество не поступит в ячейку' Посредством автоматической подстройки длин волн возбуждения и испускания, зависящих от характера детектируемого вещества могут быть выбраны оптимальные условия для каждого отдельного соединения во время хроматографического разделения.
Последовательное соединение детектора на диодной матрице (ДМД) и ФЛД является чрезвычайно мощным инструментом для надежного и чувствительного детектирования ПАУ в про6ax воды. Чрезвычайно высокая чувствительность флуоресцентного детектирования стимулировала разработку надежных автоматических реакторов для перевода нефлуоресцирующих веществ в форму флуоресцирующих производных и последующего их детектирования с помощью ФЛД.
Из других детекторов, используемых в ВЭЖХ, следует отметить электрохимический (ЭХД) и масс-спектрометрическии (МСД) детекторы. Оснащенный компьютером ЭХД применяют для обнаружения и количественного определения токсичных веществ, которые легко окисляются или восстанавливаются. К ним относятся фенолы, меркаптаны, амины, ароматические нитро- и галогенпроизводные, альдегиды, кетоны и, особенно, бензидины.
В свое время масс-спектрометрия революционизировала аналитическую химию, став одним из наиболее важных инструментов природоохранительной лаборатории. Однако, если масс-спектрометр может быть непосредственно соединен с газовым хроматографом, то соединение его с жидкостным хроматографом требует специального интерфейса Последнее обстоятельство пока ограничивает широкое применение комбинации ВЭЖХ/МСД в экологической аналитической химии.
Традиционный УФ-детектор с перестраиваемой длиной волны для ВЭЖХ по существу представляет собой высокочувствительный УФ-спектрометр с проточной микроячейкой, который регистрирует оптическую плотность раствора при данной длине волны. В настоящее время наиболее популярна высокочувствительная запись спектров с помощью детекторов на диодной матрице. В таких детекторах линейка фотодиодов (более двухсот) постоянно регистрирует сигналы в ультрафиолетовой и видимой частях спектра (УФ-В-детекторы), обеспечивая запись в режиме сканирования. Данные, полученные одновременно на различных длинах волн, обрабатываются с помощью компьютеров, которые выделяют сигнал на оптимальной длине волны, вычитают фон и осуществляют другие операции. В частности, с появлением УФ-В-детектора на диодной матрице ВЭЖХ стала стандартным методом контроля качества природной и питьевой воды на содержание пестицидов.
|
|
|
|
|
Дата добавления: 2014-01-05; Просмотров: 3438; Нарушение авторских прав?; Мы поможем в написании вашей работы!