КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Химическая кинетика и Катализ. Химическое равновесие
|
|
|
|
Как протекают реакции во времени? Закономерностями протекания процессов во времени занимается химическая кинетика. Для управления химическими процессами необходимо знание скоростей химических реакций и факторов, влияющих на скорость.
§ 1. Скорость химической реакции. Основным понятием химической кинетики является скорость реакции. Скоростью химической реакции называют изменение количества реагирующего вещества или образующегося продукта в единице объема за единицу времени.
Для гомогенной (однофазной) системы реакционным пространством служит объем, и скорость v может быть выражена:
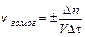 ;
; 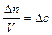 ;
; 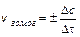 ;
; 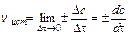 (9.1)
(9.1)
где D n – изменение количества вещества; t – время; D c – изменение концентрации. В кинетике под реагентами понимают исходные вещества, а образующиеся в ходе реакции вещества – продукты.
Изменение концентрации веществ показаны на рис. 9.1. Видно, что u для исходных веществ имеет знак (–) минус.
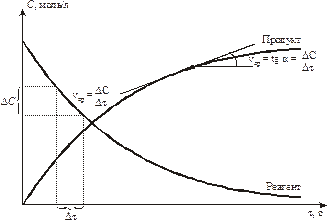
Рис. 9.1. Изменение концентраций реагентов и продуктов при химическом взаимодействии.
Для гетерогенных систем (различные фазы) реакции протекают на поверхности раздела фаз, и скорость определяется количеством вещества, вступившего
в реакцию или получившегося в результате реакции (D n) за единицу времени (Dt) на единице поверхности раздела (S):
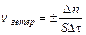 ;
; 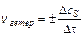 ;
; 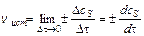 (9.2)
(9.2)
Основной закон химической кинетики – закон действующих масс (ЗДМ), высказанный в 1865 г. М.Н. Бекетовым и уточненный норвежскими химиками Гульдбергом и Вааге. Зависимость скорости химической реакции от концентрации выражается законом действующих масс: скорость простой химической реакции прямо пропорциональна концентрации реагирующих веществ, взятых в степенях, равных стехиометрическим коэффициентам в уравнении реакции (aA+bB=dD), или в математическом виде:
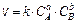 (9.3)
(9.3)
где k – константа скорости химической реакции, равная скорости химической реакции при концентрациях реагирующих веществ 1 моль/л.; a и b – частные порядки реакции, С – концентрации.
Например, для реакции 2А+В=А2В скорость определится следующим
образом:
v = k CA 2 CB (9.4)
Константа скорости не зависит от концентрации веществ, а зависит от температуры. Выше указанное отношение (9.3) справедливо только для гомогенных реакций. Для гетерогенных процессов концентрация вещества в твердой фазе не меняется, поэтому скорость определяется концентрацией газов или растворенных веществ. Так, для реакции С+О2=СО2
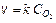 (9.5)
(9.5)
Обычно концентрацию газов заменяют парциальным давлением газа PO 2. Численное значение k при этом станет другим.
Классификация химических реакций. В химической кинетике реакции классифицируют по I. молекулярности и порядку реакции; II. по механизму протекания.
I. 1) Молекулярность определяется числом молекул, участвующим в элементарном химическом акте:
Н2 = 2Н – мономолекулярная;
CН3COOC2H5 + H2O = CН3COOH + C2Н5OH – бимолекулярная;
2NO + Cl2 = 2NOCl – тримолекулярная.
Реакции с молекулярностью большей трех не встречаются, т.к. вероятность столкновения сразу четырех и более молекул очень мала.
2) Порядок реакции – это сумма показателей степеней при концентрациях веществ в уравнении закона действующих масс. Так, реакции (9.5) – первого порядка, реакция (9.4) – третьего порядка. Иногда указывают частный порядок реакции по веществу – это степень, в которую возводится концентрация этого вещества.
II. 1) Параллельные реакции. Практически выгодным является получение перхлората калия, который является одним из компонентов ракетного топлива, поэтому изучение скорости и факторов влияющих на скорость химических реакций позволяет управлять параллельными реакциями и достигнуть v 1 < v 2.
2KOH + Cl2  KClO + KCl, 6KOH + Cl2
KClO + KCl, 6KOH + Cl2  KClO + 5KCl
KClO + 5KCl
2) Последовательные реакции А В
В С
С D. Чтобы управлять процессом, необходимо выявить стадию, у которой скорость реакции минимальна, то есть скорость реакции, которая является лимитирующей для всего процесса. Затем, подбирая условия реакции, увеличивать скорость лимитирующей стадии, тем самым будет увеличиваться скорость всего процесса.
D. Чтобы управлять процессом, необходимо выявить стадию, у которой скорость реакции минимальна, то есть скорость реакции, которая является лимитирующей для всего процесса. Затем, подбирая условия реакции, увеличивать скорость лимитирующей стадии, тем самым будет увеличиваться скорость всего процесса.
3) Сопряженные реакции. Одна реакция идет только в том случае, если идет вторая.
§ 2. Уравнение Аррениуса. Скорость химической реакции зависит от температуры. При повышении температуры скорость большинства химической реакций увеличивается. Вант-Гофф показал, что при повышении температуры на 10 град., скорость реакции увеличивается в 2-4 раза (правило Вант-Гоффа):
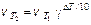 (9.6)
(9.6)
Температурный коэффициент реакции g равен отношению скоростей реакции, когда Т2–Т1=10 град. и показывает, во сколько раз увеличивается скорость реакции при повышении температуры на 10 град. Но уравнение (9.6) лишь приблизительно оценивает зависимость скорости реакции и константы скорости от температуры. Функциональную зависимость константы скорости реакции от температуры установил Аррениус (9.7).
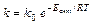 (9.7)
(9.7)
где k 0 – начальная скорость или предэкспоненциальный множитель – А. Графически величину Е а по Аррениусу можно представить на рис. 9.2.
В логарифмической форме это уравнение для констант скоростей преобразуется:
ln k = – Ea / RT + ln k 0 (9.8)
Видно, что зависимость константы скорости реакции от температуры линейна, следовательно можно определить энергию активации по тангенсу угла наклона прямой и k 0 – по отрезку, отсекаемому прямой по оси ординат, когда 1/Т→0. Энергию активации легко рассчитать, если известны константы скорости при разных температурах.
Все молекулы, запас энергии, которых не ниже энергетического барьера называются активными и они находятся в состоянии активированного комплекса. В состоянии активированного комплекса нет уже исходных веществ, но и еще нет продуктов реакции (старые связи еще не до конца разорвались, а новые
не образовались). Энергия такой системы максимальна, а значит, активированный комплекс неустойчив.
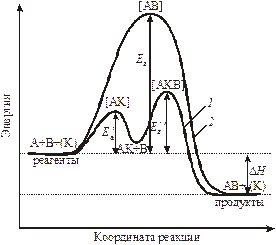
Рис. 9.2. Энергетический профиль реакции: 1 – в присутствии катализатора; 2 – без катализатора. Под координатой реакции понимается многопараметровая функция, зависящая от времени, пространственных координат молекул реагентов и др.
Разница в энергиях между исходным и переходным состоянием равна энергии активации. То есть, активные молекулы должны обладать энергией активации для осуществления взаимодействия для того, чтобы произошло ослабление связей в исходных веществах, для преодоления отталкивания между электронами в сближающихся молекулах, которое мешает их столкновению. Следовательно, энергия активации является одним из параметров, характеризующих скорость процесса. Чем больше энергия активации, тем меньше скорость процесса. Если Еа больше 1000 кДж/моль, то реакция не протекает при комнатной температуре, такие реакции идут при особых условиях, высоких Т и Р. Энергия активации в первую очередь зависит от природы реагирующих частиц. Существуют реакции, у которых Еакт = 0 – это туннельные реакции, протекающие, как правило, при низких температурах.
§ 3. Влияние катализаторов на скорость химической реакции. Катализаторами называются вещества, увеличивающие скорость реакции, но не расходующиеся в результате ее протекания. Катализаторы обычно участвуют в образовании промежуточных соединений, но в конце реакции полностью регенерируются. Наряду с катализаторами существуют ингибиторы, замедляющие скорость реакции. Катализ – наиболее эффективный метод интенсификации промышленных химических процессов. С точки зрения закона Аррениуса катализаторы (К) снижают энергетический барьер, а ингибиторы увеличивают (рис. 9.2).
1) Катализ применим только тогда, когда свободная энергия Гиббса данной реакции отрицательна (DG<0).
2) В присутствии катализатора изменяется механизм химической реакции, она протекает через новые стадии, характеризующиеся небольшими значениями Еа.
3) При катализе не изменяется тепловой эффект реакции.
4) Если реакция не обратима, то катализатор не влияет на положение равновесия.
5) Катализаторы обычно действуют избирательно, селективно. Одним из универсальных катализаторов является вода.
Активность того или иного катализатора зависит от многих факторов. Активность катализатора может быть повышена специальными добавками – промоторами. Если катализатор и реагирующие вещества образуют одну фазу, то катализ называют гомогенным, если две фазы – гетерогенным. Для объяснения гомогенного катализа используют теорию промежуточных соединений. Согласно этой теории катализатор взаимодействует с одним из исходных веществ с образованием промежуточных соединений, при этом понижается энергия активации и растет скорость реакции. А+В=AB; А+К=[АК]; [АК]+В=AB+К.
Для своего прохождения реакция должна преодолеть «энергетический барьер» – энергию активации и чем меньше барьер, тем больше скорость реакции. Таким образом, скорость реакции определяется числом активных молекул и величиной энергии активации, последняя изменяется в пределах 40-350 кДж.
§ 4. Химическое равновесие. Химические реакции бывают необратимые (односторонние) и обратимые (двухсторонние) или равновесные. Необратимые реакции протекают только в одном направлении до полного превращения хотя бы одного из реагирующих веществ. Примером необратимой реакции может служить реакция разложения бертолетовой соли:
2KClO3 = 2KCl + 3O2
Обратимые реакции могут протекать как в прямом, так и в обратном направлении, в связи с этим ни одно из реагирующих веществ не расходуется полностью. Так, например, следуя рис. 9.1, в состоянии равновесия имеет место определенное количество как реагента, так и продукта. Например, реакция синтеза аммиака является обратимой:
3Н2 + N2 ⇄ 2NН3
Она может протекать как в сторону образования аммиака (прямой процесс), так и в сторону реагентов – молекулярного азота и водорода (обратный процесс). Знак обратимости реакции – стрелки с противоположными направлениями: ⇄.
Выразим для обратимого процесса две скорости прямой (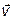 ) и обратной реакции (
) и обратной реакции (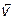 ) по закону действующих масс:
) по закону действующих масс:
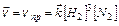 (9.9)
(9.9)
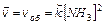 (9.10)
(9.10)
Когда скорости прямой и обратной реакций становятся равными, наступает химическое равновесие. Комбинируя уравнения 9.9, 9.10, получим:
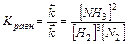 (9.11)
(9.11)
Это уравнение отвечает кинетическому условию равновесия. Химическое равновесие называют динамическим. Этим подчеркивается, что при равновесии протекают как прямая, так и обратная реакции, но их скорости равны, а устанавливается оно с течением времени.
Кравн – константа равновесия является количественной мерой, характеризующей реакцию в состоянии химического равновесия. Закон действующих масс для обратимого процесса можно сформулировать: В состоянии химического равновесия отношение произведения концентраций продуктов реакции к произведению концентраций исходных веществ есть величина постоянная. При этом концентрация каждого вещества берется в степени его стехиометрического коэффициента в уравнении реакции. Значения некоторых Кравн приведены в «Практикуме».
Система находится в состоянии равновесия до тех пор, пока внешние условия сохраняются постоянными. Если же условия изменятся, то система выйдет из равновесия. При этом скорости прямого и обратного процессов изменяются неодинаково, и будет протекать преимущественно только один процесс (прямой или обратный), т.е. произошло смещение равновесия. Оно происходит под действием таких факторов: изменение концентрации веществ, температуры, давления и подчиняется универсальному принципу Ле-Шателье: Всякое внешнее воздействие на равновесную химическую систему вызывает в ней процесс, приводящий к уменьшению эффекта внешнего воздействия, который часто называют «третьим законом Ньютона в химии» и имеющий универсальный характер в природе.
Влияние изменения концентрации веществ на смещение равновесия. По принципу Ле-Шателье увеличение концентрации исходных веществ сместит равновесие в сторону той реакции, при которой происходит уменьшение количества исходных веществ, т.е. в сторону превращения исходных веществ в продукт реакции.
Наоборот, увеличение концентрации продуктов реакции сместит равновесие в сторону образования исходных веществ. Уменьшение же концентрации продуктов реакции сместит равновесие в сторону образования продуктов реакции и концентрация их возрастет.
Обобщая, можно сделать вывод, что при увеличении концентрации какого-либо из веществ равновесие смещается в сторону расхода этого вещества;
при уменьшении концентрации какого-либо вещества равновесие смещается в сторону образования этого вещества.
Равновесие подавляющего большинства реакций смещается при изменении температуры. Фактором, определяющим направление смещения равновесия, является знак теплового эффекта реакции. Из принципа Ле-Шателье следует, что при повышении температуры равновесие смещается в сторону эндотермической реакции, а при охлаждении – в сторону экзотермической реакции.
В рассматриваемом случае синтез аммиака представляет собой экзотермический процесс: D H =–92 кДж/моль. Поэтому при повышении температуры равновесие смещается в сторону обратной реакции, разложения аммиака.
В соответствии с принципом Ле-Шателье при увеличении давления в системе путем сжатия равновесие сдвигается в сторону уменьшения числа молей газа; при уменьшении давления равновесие сдвигается в сторону возрастания числа молей газов.
Например, при синтезе аммиака при протекании прямого процесса из четырех молей газа образуется два моля газа, значит число молей газа уменьшается в два раза, что приводит к уменьшению давления в сосуде. При обратном процессе число молей газа наоборот возрастает, следовательно, возрастает и давление. Отсюда, вывод: синтез аммиака нужно вести при высоких давлениях. В том случае, когда реакция протекает без изменения числа молей газа, равновесие при изменении давления не нарушается. Напомним, что термодинамическим критерием химического равновесия является DG=0, что особенно наглядно проявляется при фазовых переходах.
Термодинамические и кинетические критерии химического равновесия связаны уравнением:
D G = – RT ln Kравн (9.12).
Из уравнения видно, что высоким отрицательным значениям DG отвечает большое значение константы равновесия (Kp), это значит, что в равновесной смеси преобладают продукты реакции. Если же DG имеет большое положительное значение (Kp), то в равновесной смеси преобладают исходные вещества. Уравнения (9.9) позволяют по величине DG вычислить константу равновесия (Кравн), а затем и равновесные концентрации или парциальные давления реагентов.
Таким образом, количественно химическое равновесие оценивается термодинамическим критерием (DG) и кинетическим критерием (равенство скоростей прямой и обратной реакций, Kp).
Лекция 10
Растворы
§ 1. Определение растворов. Растворимость. Способы выражения концентрации. Для того чтобы между двумя веществами произошла химическая реакция, их молекулы должны вступить в контакт друг с другом. Так, если измельчить твердые вещества в порошок, то реакции между ними ускорятся во много раз. Растворяя эти вещества, мы дробим их до наименьших возможных частиц – молекул или ионов, поэтому большинство химических реакций оказывается выгодным проводить в растворах. Реакции, протекающие в растворах, имеют важное значение для биохимии, химической технологии и т.д. Современное учение о растворах создавалось на протяжении примерно двухсот лет, в него внесли вклад такие знаменитые ученые, как М. Фарадей, Гельмгольц, Д.И. Менделеев, Аррениус, Вант-Гофф, Оствальд, Каблуков и другие. Современное определение понятия раствор следующее:
Раствором называется гомогенная многокомпонентная химическая система, состав которой в определенных пределах может варьироваться (быть переменным) без качественного изменения свойств.
Это определение требует некоторых комментариев. Гомогенная – значит, ее свойства однородны по всему объему, т.е. отсутствуют границы раздела фаз; многокомпонентная – означает совокупность нескольких веществ: растворителя и одного или нескольких растворенных веществ – компонентов. Химическая система переменного состава означает взаимодействие растворителя и компонентов с образованием химических соединений переменного состава.
Химическое взаимодействие растворителя с компонентами называется сольватацией, а в случае растворителя воды – гидратацией. Процесс этот сопровождается поглощением или выделением тепла, как и в других химических реакциях. Образующиеся сольваты и гидраты в растворе в зависимости от концентрации, температуры, давления и других факторов имеют переменный состав в отличии от исходных реагентов: растворителя и компонентов. Например, H2O и C2H5OH – соединения постоянного состава образуют сольваты {(H2O)x× (C2H5OH)y}, где x и y – целые числа (1,2,3) – соединения переменного состава. Растворы как бы занимают промежуточное положение между смесями и химическими соединениями. Растворы обладают определенной структурой, особенно для ближайшего окружения сольватов.
Важной количественной характеристикой растворов являются концентрация и растворимость. Под концентрацией понимается количество растворенного вещества в объеме раствора (растворителя). Под растворимостью понимается максимально возможное количество растворенного вещества в объеме (массе) растворителя до появления осадка (гетерогенная система, и есть граница раздела фаз). Естественно, они зависят от температуры и других внешних факторов. Растворы могут быть твердыми, жидкими и газообразными. Они также классифицируются на растворы неэлектролитов и электролитов, о которых будет идти речь в следующих лекциях.
Как правило, растворимость твердых веществ повышается с ростом температуры (хотя есть исключения) и мало зависит от изменения внешнего давления. Растворимость газов также варьирует в широких пределах (от 0,2 объема на
1 объем воды у гелия до 800 объемов у аммиака), однако понижается с ростом температуры и повышается с ростом парциального давления газа.
В случае ограниченной растворимости можно выделить растворы ненасыщенные, содержание растворенного вещества в которых меньше, чем максимально возможное при данных условиях; насыщенные, в которых данное вещество при данных условиях больше не растворяется, и пересыщенные, содержание растворенного вещества в которых больше, чем в насыщенном растворе при данных условиях (пересыщенный раствор можно получить, например, быстрым охлаждением раствора вещества, растворимость которого значительно повышается с ростом температуры). Пересыщенные растворы метастабильны, т.е. способны под действием внешних факторов, а иногда и самопроизвольно, переходить в устойчивое состояние (насыщенный раствор + избыток растворенного вещества в отдельной фазе).
Способы выражения концентрации растворов даны в «Практикуме». Значения растворимостей различных веществ в различных растворителях табулированы в многочисленных химических справочниках.
§ 2. Термодинамические свойства растворов. Для растворов характерны несколько законов. К ним относятся: понижение давления пара растворителя, повышение температуры кипения и понижение температуры замерзания раствора, осмотическое давление и другие, связывающие концентрацию, растворимость с физическими параметрами раствора, как химической системы.
Величина относительного понижения давления пара (депрессии) над раствором по сравнению с чистым растворителем пропорциональна концентрации растворенного вещества (закон Рауля):
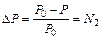 , (10.1)
, (10.1)
где P 0 – давление пара над чистым растворителем, P — давление пара над раствором, N 2 – мольная доля растворенного вещества.
Физическую природу этого явления можно пояснить следующим образом. В замкнутой системе, состоящей из жидкого растворителя и паров над ним, при постоянных условиях устанавливается динамическое равновесие между жидкой и газообразной фазой: количество молекул, перешедших из газообразной фазы в жидкую за данный промежуток времени, равно количеству молекул, перешедших из жидкой фазы в газообразную. Первая величина зависит от содержания молекул растворителя в единице объема газовой фазы, вторая — от количества молекул растворителя на единицу поверхности раздела фаз. Добавление
нелетучего растворенного вещества уменьшает только вторую величину (часть поверхности занята молекулами растворенного вещества), поэтому положение равновесия должно сместиться в сторону уменьшения давления пара растворителя. Т.е. давление пара воды над морской водой меньше чем над пресной. У закона Рауля есть два следствия, связанные с кипением и замерзанием (отверждением) растворов по сравнению с растворителем.
Повышение температуры кипения растворов, по сравнению с растворителем называется эбулиоскопией, а математически выражается формулой:
D Tкип = KE × Cm, (10.2)
где KE – эбулиоскопическая постоянная для данного растворителя, Сm – моляльная концентрация. Аналогично для замерзания растворов – криоскопия и уравнения для температуры замерзания раствора по сравнению с чистым растворителем и имеет вид:
D Tзам = KE × Cm, (10.3)
где КК – криоскопическая постоянная. Методами эбулиоскопии и криоскопии можно, например, определить молекулярную массу неизвестного вещества. Как известно, если к g 1 г растворителя добавить g 2 г растворенного вещества, то моляльная концентрация будет равна:
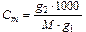 , (10.4)
, (10.4)
где M – молекулярная масса растворенного вещества.
Тогда из уравнений (10.2)-(10.4) следует, что
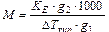 или
или 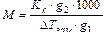 (105)
(105)
Наглядно следствия из закона Рауля объясняются изменениями на диаграмме состояния воды и раствора (рис. 10.1).
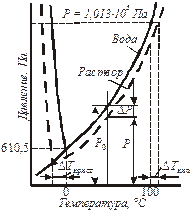
| Рис. 10.1. Фазовая диаграмма воды и раствора при постоянной концентрации. |
При пониженном по сравнению с чистым растворителем давлении паров над раствором «ветвь» граница раздела фаз понижается с увеличением концентрации растворенного вещества. При этом раствор кипит при большей температуре, чем растворитель, а замерзает при более низкой.
У эбулиоскопии и криоскопии очень много прикладных значений (посыпание солью снега зимой, охлаждающие смеси, высокотемпературные жидкости и т.д.).
У закона Рауля есть «родственник» – закон Генри: растворимость газов в жидкости пропорционально давлению:
C = k×p, (10.6)
где k – константа Генри, p – давление. Он наглядно иллюстрируется на примерах: сифон, газированная вода, наличием кислорода на больших водных глубинах, водолазная техника («кессонная болезнь») и т.д.
И наконец рассмотрим еще один закон – закон осмотического давления Вант-Гоффа.
P p = C×R×T, (10.7)
где P p – осмотическое давление, C – молярная концентрация, R – газовая постоянная, T – температура; напоминающий уравнение Клайперона – Менделеева, но связанный с явлением осмоса: давление растворителя над полупроницаемой мембраной выше, чем в растворе (с другой стороны мембраны). Не вдаваясь в суть явления осмоса, отметим, что под полупроницаемой понимается мембрана, проницаемая для молекул растворителя, но задерживающая молекулы растворенного вещества. Закон также имеет большое прикладное значение в природе и технике: рост растений, деревьев, очистка жидкостей и т.д.
Перечисленные термодинамические законы являются общими для всех растворов нелетучих веществ.
Лекция 11, 12
РАСТВОРЫ ЭЛЕКТРОЛИТОВ.
электролитическАЯ диссоциациЯ
Вещества, растворы (или расплавы) которых проводят электрический ток, называются электролитами, для которых Фарадей в начале XIX в. ввел понятие катион и анион и установил законы электролиза. Носителями электричества в растворе являются катионы и анионы, т.е. атомы и молекулы несущие положительный и отрицательный заряд, по Фарадею возникающие в процессе прохождения электрического тока. Лишь спустя почти полвека великий Сванте Аррениус установил, что все как раз наоборот: ионы (катионы, анионы) образуются в растворах электролитов всегда вследствие электролитической диссоциации полярных молекул и ионных кристаллов.
§ 1. Основные положения теории электролитической диссоциации. При растворении солей, кислот и оснований в воде происходит диссоциация этих веществ с образованием электрически заряженных частиц – катионов и анионов; электрическая проводимость водных растворов солей, кислот и оснований пропорциональна общей концентрации ионов в растворе. Таким образом, ионы самопроизвольно, изначально существуют в растворе электролитов, так что в целом раствор электронейтрален, а при наложении разности потенциалов проводит электрический ток. Аррениус ввел представление об электролитической диссоциации для объяснения природы изотоническогоё коэффициента – i, эмпирически в веденного Вант-Гоффом для выполнения термодинамических законов растворов электролитов, т.е. для законов Рауля, эбулиоскопии, криоскопии, Вант-Гоффа (10.1-10.5) он предложил ввести изотонический коэффициент – множитель концентрации. Так что, например, для осмотического давления следует писать
p = iCRT, (11.1)
С введением изотонического коэффициента эффективная концентрация электролита увеличивалась. По Аррениусу она увеличивалась за счет диссоциации – распада молекулы на несколько ионов.
Аррениус ввел понятие – степень электролитической диссоциации (a) как отношение числа молекул, продиссоциированных на ионы к общему числу молекул в растворе. Она меняется от 0 до 1, или от 0 до 100%.
Вспомните слабые, сильные электролиты, теперь они характеризуются a, которая связана с изотоническим коэффициентом следующим соотношением:
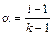 , (11.2)
, (11.2)
где i = 1 для растворов неэлектролитов, i > 1 для растворов электролитов. С уменьшением концентрации i увеличивается 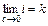 ; k – целое число ионов в результате полной диссоциации, для бинарных электролитов (KCl, HCl) k = 2, для H3PO4: k =4, i <4 и т.д.
; k – целое число ионов в результате полной диссоциации, для бинарных электролитов (KCl, HCl) k = 2, для H3PO4: k =4, i <4 и т.д.
Таким образом, в растворах электролитов имеет место гидратация и сольватация (Менделеев) и электролитическая диссоциация (Аррениус) как два единых процесса растворения электролитов в растворе («две стороны одной медали»), что и ввел в теорию и практику Каблуков.
Электролитическая диссоциация вызывается взаимодействием полярных молекул растворителя с частицами растворяемого вещества. Это взаимодействие приводит к поляризации связей. Происходит образование ионов за счет «ослабления» и разрыва связей в молекулах растворяемого вещества. Переход ионов в раствор сопровождается их гидратацией:
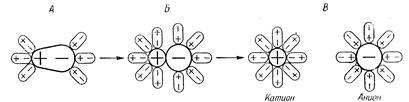
Рис. 11.1. Схема процессов гидратации и диссоциации полярной молекулы электролита типа HCl (разрыв ионогенной полярной ковалентной связи с ее переходом в ионную связь).
Кратко остановимся на водных растворах электролитов, основными участниками которых являются вода, кислоты, щелочи, соли и образуемые ими гидраты ионов (аквакомплексы), комплексные соединения.
Особенностью водных растворов электролитов является ион водорода (протон), имеющий малый размер (10–4 от размера атома), и образующий при гидратации ион гидроксония – Н3О+. Новая ковалентная связь образуется по донорно-акцепторному механизму за счет свободной электронной пары кислорода (донора) и вакантной орбитали протона (акцептора).
Характер ионов, образующихся при диссоциации различных электролитов, естественно, должен быть различным. В молекулах солей диссоциация всегда приводит к образованию катионов металла и анионов кислотного остатка. Кислотами являются электролиты, которые диссоциируют с образованием ионов Н+. Сильные кислоты (HCl, HBr, HI, HNO3, H2SO4) диссоциируют практически полностью, у слабых кислот диссоциирована лишь часть молекул. Основание определяют как электролит, диссоциирующий с образованием ионов ОН–. Сильные основания (NaOH, KOH) диссоциируют практически полностью, у слабых электролитов диссоциирована лишь некоторая доля молекул.
Существуют электролиты, которые могут диссоциировать как кислоты и как основания. Такие электролиты называются амфотерными электролитами (амфолитами). Амфотерность электролитов объясняется малым различием прочности связей Me–O и H–O. Примером амфотерного электролита может быть гидроксид цинка:
2H++ZnO22– ⇄ H++HZnO2– ⇄ Zn(OH)2 ⇄ZnOH++OH– ⇄Zn2++2OH–
При взаимодействии с азотной кислотой
Zn(OH)2 + 2HNO3 = Zn(NO3)2 + 2H2O
при взаимодействии с гидроксидом калия
Zn(OH)2 + 2KOH = K2ZnO2 + 2H2O
В растворах слабых электролитов процесс диссоциации протекает обратимо и, следовательно, к нему может быть применен закон действия масс, например:
CH3COOH ⇄ CH3COO–+ H+
Константа равновесия Kp будет равна:
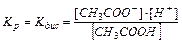 (11.3).
(11.3).
Константа равновесия для процесса диссоциации называется константой диссоциации Kдис.
Установлено, что многоосновные кислоты отщепляют ион водорода не сразу, а постепенно. Например,
1) H3PO4 ⇄ H++H2PO4– K1=7,11×10–3
2) H2PO4 ⇄ H++HPO42– K2=6,34×10–8
3) HPO4 ⇄ H++PO43– K3=1,26×10–12
Каждая ступень характеризуется своей константой диссоциации, но первая ступень идет легче, поэтому К1 больше следующей и т.д.
Константа диссоциации представляет собой важную характеристику слабых электролитов, так как указывает на прочность их молекул в данном растворе. Чем меньше константа диссоциации в данном растворителе, тем слабее диссоциирует электролит и тем устойчивее его молекулы.
Константа электролитической диссоциации зависит лишь от природы электролита и температуры. Между константой и степенью электролитической диссоциации существует количественная связь. Действительно, пусть в рассмотренном процессе общая концентрация растворенного вещества КА равна C, а степень диссоциации равна a. Тогда [ K +] = [ A –] = a× C и соответственно концентрация недиссоциированных частиц [ KA ] = (1–a)× C. Подставив эти значения в выражение для константы диссоциации, получим известный закон разбавления Оствальда:
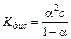 (11.4)
(11.4)
Это соотношение называется законом разбавления Оствальда. Для слабых электролитов, когда a <<1, Кдис ≈ a2 C. Отсюда
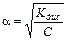 или
или 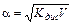 , (11.5)
, (11.5)
где V =1/ C – разбавление. Из формулы следует, что при разбавлении раствора в 100 раз степень диссоциации возрастет в 10 раз.
Электропроводность растворов электролитов зависит от их концентраций в растворе. Величина, характеризующая данный раствор, называется удельной электропроводностью c
c = 1 / r (11.6)
где r – удельное электрическое сопротивление.
С разбавлением раствора она уменьшается, вследствие уменьшения концентрации, но в тоже время увеличивается степень диссоциации.
§ 2. Сильные электролиты. Активность. Согласно теории диссоциации, сильные электролиты практически полностью диссоциируют не только в разбавленных растворах, но и в растворах любой концентрации. Другими словами, в любых растворах сильных электролитов степень их диссоциации a =1 (100%).
Таким образом, считается, что в растворах сильных электролитов нет недиссоциированных молекул, поэтому понятие константы диссоциации к ним не применимо (знаменатель выражения вида (11.3) равен нулю).
При определенных условиях, например, когда растворитель обладает малой диэлектрической проницаемостью, создаются условия для электростатического взаимодействия сольватированных ионов противоположного знака. При этом последние подходят друг к другу на близкое расстояние и образуют так называемую ионную пару – сложный агрегат, состоящий из двух противоположно заряженных ионов, окруженных молекулами растворителя, в котором электрические заряды взаимно компенсированы.
При повышении концентрации раствора расстояния между ионами сокращаются, что усиливает межионное взаимодействие. Вследствие этого экспериментально определяемые свойства растворов сильных электролитов, зависящие от общего количества частиц в растворе, оказываются меньше рассчитанных в предположении полной диссоциации.
Чтобы можно было пользоваться простыми соотношениями идеальных растворов для описания поведения реальных растворов, Льюис (1907) ввел формальное представление об эффективной концентрации – активности. Активность связана с истинной концентрацией растворенного вещества соотношением
a = f × C, (11.7)
где a – активность, f – коэффициент активности, C – концентрация.
Активность выражается в тех же единицах, что и концентрация, поскольку коэффициент активности – величина безразмерная. Он характеризует степень отклонения свойств данного раствора от свойств идеального раствора. Для бесконечно разбавленных растворов электролитов, где практически отсутствует взаимодействие ионов, активность становится равной концентрации и коэффициент активности равен единице.
Так как коэффициенты активности учитывают также степень диссоциации электролита в растворе, их применяют и при описании свойств растворов слабых электролитов. Коэффициенты активности вычисляют по экспериментальным данным. Для этого измеряют какое-либо из свойств раствора (например, электропроводность, температуру кипения или замерзания) и определяют коэффициент активности как частное от деления экспериментально полученной величины на теоретически рассчитанную по законам идеальных растворов:
Таблица 11.1
Коэффициенты активности некоторых электролитов в растворах при 298 К
| Концентрация, | f ± для электролитов | ||||||
| NaCl | KCl | NaOH | KOH | HCl | H2SO4 | CaCl2 | |
| 0,001 0,01 0,1 0,5 1,0 2,0 5,0 | 0,965 0,874 0,778 0,681 0,657 0,668 0,874 | 0,966 0,901 0,769 0,651 0,607 0,576 — | 0,966 0,900 0,776 0,693 0,679 0,700 1,060 | 0,966 0,900 0,766 0,712 0,735 0,863 1,670 | 0,966 0,904 0,796 0,758 0,809 1,010 2,380 | 0,830 0,544 0,265 0,156 0,132 0,128 0,208 | 0,840 0,850 0,518 0,448 0,500 0,792 0,890 |
В области разбавленных растворов (ниже 0,1 моль/л) коэффициенты активности зависят главным образом от концентрации и заряда ионов, присутствующих в растворе, и мало зависят от природы растворенных веществ. Эта закономерность известна в теории растворов под названием «правила ионной силы», учитывающее электростатическое взаимодействие ионов и диполей. Согласно этому правилу ионы одинакового заряда независимо от их природы в разбавленных растворах с одинаковой ионной силой имеют равные коэффициенты активности. Ионной силой раствора (I) называется полусумма произведений концентрации всех ионов, на квадрат их заряда (z):
 (11.8)
(11.8)
где i – порядковый номер иона.
3. Водородный показатель. Индикаторы. Вода служит не только наиболее распространенным растворителем для многих электролитов, но и сама является идеальным амфолитом. В соответствии с равновесием
H2O ⇄ H+ + OH–
в чистой воде присутствуют катионы водорода и гидроксид-анионы в строго эквивалентных количествах. Константа диссоциации воды
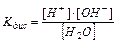 (11.9)
(11.9)
имеет значение в стандартных условиях – 1,8×10–16 т.е. вода диссоциирована в очень малой степени. Так как вода – очень слабый электролит, то концентрация недиссоциированных молекул может быть принята равной общему количеству молей в 1 л воды, т.е. [Н2O] = 1000/18 = 55,56 моль/л. Тогда Kдис ×[ H2O ] = [ H+ ]×[ OH– ] или [ H+ ]×[ OH– ] = 1,8×10–16× 55,56 = 10–14 (константа воды).
Величина [ H+ ]×[ OH– ] = 10–14 называется ионным произведением воды –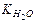 , Kw. Так как в воде концентрации гидратированных ионов равны, то [ H+ ] = [ OH– ] =
, Kw. Так как в воде концентрации гидратированных ионов равны, то [ H+ ] = [ OH– ] = 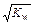 = 10–7 моль/л.
= 10–7 моль/л.
Константа равновесия Kw увеличивается с ростом температуры. При добавлении кислоты концентрация ионов водорода увеличивается и соответственно уменьшается концентрация гидроксид-ионов, поскольку при данной температуре ионное произведение воды – величина постоянная. При добавлении щелочи наблюдается обратная картина. Таким образом, концентрация ионов водорода в растворе может служить мерой кислотности или щелочности среды. В кислых растворах [H+] > 10–7, в щелочных [H+] < 10–7. Вводится значение десятичного логарифма концентрации водородных ионов с обратным знаком, которое называют водородным показателем pH:
pH = –lg[ H+ ] (11.10)
Тогда для нейтральной среды pH = 7, для кислых растворов рН < 7, а для щелочных рН > 7. Аналогичным образом реакция среды может быть охарактеризована так называемым гидроксильным показателем:
pOH = –lg[O H– ](11.11)
Для воды рН = рОН = 7, а изменение рОН в кислых и щелочных растворах противоположно изменению рН. Прологарифмировав ионное произведение воды, получим
lg[ H+ ] + lg[O H– ] = –14 (11.12)
Взяв логарифмы со знаком «–», получим соотношение рН+рОН=14, Водородный показатель удобно представить в виде шкалы (рис. 11.2).
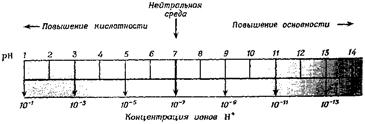
Рис. 11.2. Шкала pH.
Для определения рН используют так называемые кислотно-основные индикаторы – вещества, меняющие свой цвет в зависимости от относительной концентрации ионов H+ и ОН–. Индикаторы представляют собой слабые органические кислоты или основания. Одним из наиболее известных индикаторов является лакмус, окрашивающийся при избытке Н+ (т.е. в кислой среде) в красный цвет, при избытке ОН– (т.е. в щелочной среде) – в синий и имеющий в нейтральной среде фиолетовую окраску за счет равновесия: HInd ⇄ H+ + Ind–.
§ 4. Гидролиз. Равновесие с участием малорастворимых веществ. Наряду с электролитической диссоциацией и гидратацией (напомним, что это процесс взаимодействия ионов с молекулами воды за счет донорно-акцепторных связей, как правило, с образованием гидратированных ионов) имеется еще реакция гидролиза («разложения» водой). Гидролизом называется обменная реакция разложения соли водой, в результате которой получаются слабые кислоты или основания. Реакция гидролиза обратна реакции нейтрализации
Рассмотрим важнейшие случаи гидролиза солей.
1) Соль слабой кислоты и сильного основания. Сюда относятся такие соли как Na2CO3, K2CO3, CH3COONa, KCN и т.д.
Na2CO3 + HOH ⇄ NaHCO3 + NaOH
CO32– + HOH ⇄ HCO3–+ OH–
KOH сильное основание, хорошо диссоциируется в водном растворе, а CH3COOH – слабая кислота, распадающаяся на ионы лишь в очень малой степени. Раствор приобретает щелочную реакцию, вследствие наличия в нем свободных гидроксильных ионов в концентрации, более высокой, чем H+ ионов
[OH–]>[H+], pH>7.
2) Соль слабого основания и сильной кислоты. Сюда относятся NH4Cl, NH4NO3, AlCl3, CuSO4 и т.д.
NH4NO3 + HOH ⇄NH4OH + HNO3
NH4+ + HOH ⇄ NH4OH + H+
NH4OH – слабое основание, малодиссоциирующее, HNO3 – сильная кислота, сильнодиссоциирующая. [H+]>[OH–], pH < 7.
3) Соль образованная слабым основанием и слабой кислотой. Сюда относятся NH4CN, Al2S3,(CH3COO)3Fe
NH4CN + HOH ⇄ NH4OH+HCN
Раствор приобретает слабокислую, если кислота сильнее основания или слабощелочную, если основание сильнее кислоты, реакцию.
4) Соль, образованная сильным основанием и сильной кислотой. Она не гидролизуется, т.к. обратная гидролизу реакция нейтрализации практически необратима, т.е. протекает да конца.
Показателем глубины протекания процесса гидролиза служит степень гидролиза
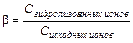 (11.13)
(11.13)
Гидролиз соли, образованной слабой кислотой HA и сильным основанием, характеризуется константой гидролиза KГ:
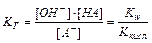 (11.14)
(11.14)
Kw – ионное произведение воды.
Аналогично для соли слабого основания MOH и сильной кислоты:
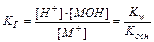 (11.15)
(11.15)
Произведение растворимости. В насыщенном растворе малорастворимого сильного электролита устанавливается равновесие между осадком (тв. фазой) электролита и ионами электролита в растворе, например:
BaSO4 (тв.) ⇄Ba2+ (р-р) + SO42– (р-р)
Т.к. в растворах электролитов состояние ионов определяется их активностями, то константа равновесия процесса выразится уравнением
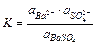 (11.16)
(11.16)
Активность сульфата бария является при данной температуре константой. Следовательно, произведение активностей ионов Ba2+ и SO42– представляют собой постоянную величину, называемую произведением растворимости и обозначаемую ПР:
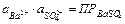 (11.17)
(11.17)
Произведение активностей ионов малорастворимого электролита, содержащихся в его насыщенном растворе (произведение растворимости), есть величина постоянная при данной температуре.
Если электролит очень мало растворим, то ионная сила его насыщенного раствора близка к нулю, а коэффициент активности ионов мало отличается от единицы. В подобных случаях произведение активностей ионов в выражении можно заменить произведением их концентраций
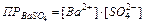 ,
, 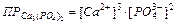
При увеличении концентрации одного из ионов электролита в его насыщенном растворе произведение концентраций ионов электролита становится больше ПР. При этом равновесие между твердой фазой и раствором смещается в сторону образования осадка. Таким образом, условием образования осадка является превышение произведения концентраций ионов малорастворимого электролита над его произведением растворимости.
Если в насыщенном растворе электролита уменьшится концентрация одного из ионов, произведение концентраций ионов будет меньше значения ПР, раствор станет насыщенном, а равновесие между жидкой фазой и осадком смещается в сторону растворения осадка. Следовательно, растворение осадка малорастворимого электролита происходит при условии, что произведения концентраций его ионов меньше значения ПР.
§ 5. Структура воды. Водородная связь. Промежуточный характер между валентным и межмолекулярным взаимодействием имеет так называемая водородная связь. Она осуществляется между положительно заряженным протоном (поляризованным атомом водорода), химически связанным в одной молекуле, и отрицательно заряженным ионом гетероатома (поляризованным атомом фтора, кислорода, азота или хлора), принадлежащим другой молекуле. То, что подобное взаимодействие не обнаруживается у других атомов, обусловлено уникальными свойствами поляризованного атома водорода, протона – его малым размером и отсутствием электронов. Водородная связь проявляется тем сильнее, чем больше относительная электроотрицательность и меньше размер атома-партнера.
Благодаря наличию водородной связи молекулы объединяются в димеры и более сложные ассоциаты, устойчивые в растворах. Ассоциаты (или кластеры) – многомолекулярные образования могут представлять собой одномерные образования, двумерные плоские сетки и трехмерные пространственные структуры.
В кристалле льда молекулы воды расположены тетраэдрически. Каждый атом кислорода тетраэдрически связан с четырьмя атомами водорода. Создается ажурная структура, далекая от плотной упаковки. При плавлении льда водородные связи частично разрушаются. Это сближает молекулы, поэтому вода плотнее льда.
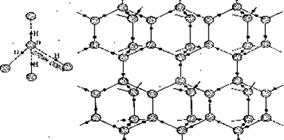
| Рис. 11.3. Структура льда с водородными связями. |
Силы, обеспечивающие возможность существования жидкой фазы, носят различный характер. Чаще всего это межмолекулярные ван-дер-ваальсовые силы взаимодействия типа диполь-диполь или диполь-индуцированный диполь, либо дисперсионные, описанные Лондоном и называемые силами взаимодействия Ван-дер-Ваальса - Лондона. Энергия последних сильно зависит от расстояния между центрами взаимодействующих частиц (она пропорциональна 1/r6), поэтому такие взаимодействия могут возникать только на очень коротких расстояниях. Энергия межмолекулярных взаимодействий, как правило, на 2-3 порядка меньше энергии обычных химических связей (2-20 кДж/моль).
Одним из интересных типов межмолекулярного взаимодействия является водородная связь, наиболее ярко и своеобразно проявляющаяся в жидкой воде, придающая особый облик ее структуре и химическим свойствам. Именно наличием водородной связи обусловлены свойства воды как универсального растворителя.
В системах с наиболее прочными водородными связями межъядерные расстояния O-Н и Н-O во фрагменте O-Н...О выравниваются. Самая короткая водородная связь обнаружена в анионе НF2–, здесь она симметрична и имеет энергию связи порядка 155-242 кДж.
Еще одним примером проявления сильной водородной связи является катион Н5О2+(напомним существование Н3О+), реализующиеся в воде в форме димера [Н2О-Н-ОН2]+, существующий в воде при определенных значениях рН. В линейном фрагменте О-Н-О реализуется водородная связь, расстояние О-О равно 0,25 нм, расстояние O-Н – 0,125 нм, энергия связи примерно 150 кДж. На этом примере можно видеть, как в результате образования Н-связей резко сокращается расстояние между взаимодействующими частицами по сравнению с обычным межмолекулярным взаимодействием. Для ассоциатов, кластеров с большим числом молекул воды водородная связь приобретает ассиметричный характер: фрагмент О-Н-О длиной 0,36 нм имеет два неравных плеча О-Н – 0,1 и 0,26 нм.
Например, в структуре льда между атомами кислорода во фрагменте O-Н...О расстояние О...О составляет 0,276 нм, что заметно отличается от 0,36 нм, однако больше, чем 0,25 нм, характерного для очень сильной водородной связи.
Какова же природа водородной связи? Долгое время ее описывали как не совсем обычное электростатическое взаимодействие.
Более глубокое проникновение в природу взаимодействий показало несостоятельность электростатического подхода. Основой электростатического взаимодействия прежде всего является ее ненаправленный характер. Поэтому появление водородной связи следовало ожидать в любом пространственном расположении двух взаимодействующих молекул. Однако опыт показывает, что водородная связь имеет строго направленный характер. И, наконец, при электростатическом подходе невозможно объяснить перераспределение валентной электронной плотности при взаимодействии молекул, которое играет существенную роль при образовании водородной связи.
Полные квантовомеханические расчеты показывают, что в линейном фрагменте О-Н···О происходит общий сдвиг валентной электронной плотности от одного кислорода к другому.
Таким образом, по своей природе водородная связь является разновидностью донорно-акцепторной связи. Особенность ее заключается в том, что электронная пара атома О молекулы Н2О непосредственно взаимодействует с атомом Н группы О-Н, через которую и осуществляется перенос заряда на другую молекулу воды. В результате такого взаимодействия происходит увеличение дипольного момента комплекса с водородной связью по сравнению с геометрической суммой дипольных моментов изолированных молекул воды. Так, эффективный дипольный момент молекул воды, связанных водородными связями, равен 2,60 Д, а свободных молекул воды – 1,85 Д.
Чем больше молекул вовлекается в образование комплекса с водородными связями, тем в большей степени происходит перераспределение электронного заряда, тем сильнее водородные связи и выше их энергия. Это означает, что в жидких системах водородные связи носят кооперативный характер, они пронизывают всю жидкую систему, вовлекая в такое взаимодействие огромное число молекул.
Итак, водородная связь есть особый вид межмолекулярного донорно-акцепторного взаимодействия, в котором принимают участие все атомы, входящие в состав молекул. Это взаимодействие носит характер направленного дрейфа электронной плотности от молекулы-донора к молекуле-акцептору. Специфика водородной связи связана с уникальной электронной структурой атома водорода, поскольку в этом случае атом молекулы-донора со своей неподеленной электронной парой может приблизиться на очень короткое расстояние к атому Н, входящему в состав молекулы акцептора. Уникальность электронной структуры атома водорода заключается в отсутствии у него заполненных электронных оболочек. Специфика водородной связи в воде и водных растворах можно охарактеризовать двумя терминами: эстафетность и динамичность, т.е. протоны обеспечивают структурную организацию кластеров за счет большой скорости «эстафетного» переноса электронной плотности. Уместно сравнить явление с «бегущей волной» зрителей на стадионе, тем самым, подчеркивая волновой характер делокализованной химической связи для большого ансамбля числа молекул.
Более детально свойства водных растворов электролитов рассмотрены в дополнении к лекции 12.
Лекция 13
Окислительно-восстановительные реакции
Другим важным типом химических реакций, протекающих, как правило, в водных растворах, являются окислительно-восстановительные реакции или реакции с переносом электрона, в которых одни реагенты теряют, а другие приобретают электроны, меняют степень окисления.
§ 1. Степень окисления. Одним из основных понятий в химии, широко использующимся при составлении уравнений ОВР, является степень окисления (с.о.) атомов.
С.о. атома (элемента) в соединении – это условный заряд, вычисленный в предположении, что соединение состоит только из ионов. При определении с.о. условно предполагают, что валентные электроны в соединении переходят к более электроотрицательным атомам, а потому соединения состоят из положительно и отрицательно заряженных ионов. В действительности же в большинстве случаев происходит не полная отдача электронов, а только смещение электронной пары от одного атома к другому. Тогда можно дать другое определение: Степень окисления – это тот электрический заряд, который возник бы на атоме, если бы электронные пары, которыми он связан с другими атомами в соединении, перешли к более электроотрицательным атомам, а электронные пары, связывающие одинаковые атомы, были бы между ними поделены.
При вычислении степеней окисления используется ряд простых правил. С.о. простых веществ, как одноатомных, так и молекулярных, равна 0 (Fe0, O ). С.о. любого простого одноатомного иона равна заряду этого иона (Na+1, Ca+1, S–2). С.о. водорода в соединениях равна +1 (
). С.о. любого простого одноатомного иона равна заряду этого иона (Na+1, Ca+1, S–2). С.о. водорода в соединениях равна +1 ( ,
,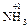 ), за исключением гидридов металлов, где она равна -1 (
), за исключением гидридов металлов, где она равна -1 ( ,
,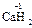 ). С.о. кислорода в соединениях равна -2 (
). С.о. кислорода в соединениях равна -2 (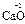 ,
,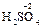 ); за исключением пероксидов, где она формально равна -1 (
); за исключением пероксидов, где она формально равна -1 (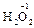 ), и фторида кислорода, где она равна +2 (
), и фторида кислорода, где она равна +2 (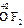 ). С.о. фтора в соединениях всегда равна -1, с.о. других галогенов (Cl, Br, I) равна -1, за исключением соединений с более электроотрицательными элементами, в которых она принимает положительные значения от +1 до +7 (
). С.о. фтора в соединениях всегда равна -1, с.о. других галогенов (Cl, Br, I) равна -1, за исключением соединений с более электроотрицательными элементами, в которых она принимает положительные значения от +1 до +7 (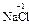 ,
, 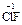 ,
,  ).
).
В ковалентных соединениях неметаллов более электроотрицательному элементу приписывается отрицательная с.о., равная заряду его наиболее распространенного аниона. Например, в CCl4 с.о. хлора -1, а углерода +4; в SF6 с.о. F
-1, а серы +6; но в CS2 с.о. серы -2, тогда с.о. углерода +4.
Алгебраическая сумма с.о. в нейтральной молекуле равна нулю, в комплексном ионе – заряду иона. Например, в NH4Cl сумма с.о. всех атомов водорода равна 4×(+1), а с.о. хлора -1, следовательно, с.о. азота должна быть -3. В сульфат-ионе SO42– сумма с.о. четырех атомов кислорода равна -8, поэтому сера должна иметь с.о. +6, чтобы полный заряд иона был равен -2. В химических реакциях должно выполняться правило сохранения алгебраической суммы с.о. всех атомов. В полном уравнении химической реакции окислительные и восстановительные процессы должны точно компенсировать друг друга.
Хотя с.о., как отмечалось выше, довольно формальное понятие, оно применяется в химии для следующих целей: во-первых, для составления уравнений ОВР, во-вторых, для предсказания окислительно-восстановительных свойств элементов в соединении.
Таблица 13.1
Степени окисления различных элементов
| Элемент | Значения степени окисления и примеры соединений |
| F | –1 (HF, KF) |
| O | –2 (H2O, CaO, CO2); –1 (H2O2); +2 (OF2) |
| N | –3 (NH3); –2(N2H4); –1 (NH2OH); +1 (N2O); +2 (NO); +3 (N2O3, HNO2); +4 (NO2); +5 (N2O5, HNO3) |
| Cl | –1 (HCl, NaCl); +1 (NaClO); +3 (NaClO2); +5 (NaClO3); +7 (Cl2O7, NaClO4) |
| Br | –1 (KBr); +1 (BrF); +3 (BrF3); +5 (KBrO3) |
| I | –1 (HI); +1 (ICl); +3 (ICl3); +5 (I2O5); +7 (IO3F, K5IO6) |
| C | –4 (CH4); +2 (CO); +4 (CO2, CCl4) |
| Si | –4 (Ca2Si); +2 (SiO); +4 (SiO2, H2SiO3, SiF4) |
| H | –1 (LiH); +1 (H2O, HCl) |
| S | –2 (H2S, FeS); +2 (Na2S2O3); +3 (Na2S2O4); +4 (SO2, Na2SO3, SF4); +6 (SO3, H2SO4, SF6) |
| Se, Te | –2 (H2Se, H2Te); +2 (SeCl2, TeCl2); +4 (SeO2, TeO2); +6 (H2SeO4, H2TeO4) |
| P | –3 (PH3); +1 (H3PO2); +3 (H3PO3); +5 (P2O5, H3PO4) |
| As, Sb | –3 (GaAs, Zn3Sb2); +3 (AsCl3, Sb2O3); +5 (H3AsO4, SbCl5) |
| Li, Na, K | +1 (NaCl) |
| Be, Mg, Ca | +2 (MgO, CaCO3) |
| Al | +3 (Al2O3, AlCl3) |
| Cr | +2 (CrCl2); +3 (Cr2O3, Cr2(SO4)3); +4 (CrO2); +6 (K2CrO4, K2Cr2O7) |
| Mn | +2 (MnSO4); +3 (Mn2(SO4)3); +4 (MnO2); +6 (K2MnO4); +7 (KMnO4) |
| Fe | +2 (FeO, FeSO4); +3 (Fe2O3, FeCl3); +4 (Na2FeO3) |
| Cu | +1 (Cu2O); +2 (CuO, CuSO4, Cu2(OH)2CO3) |
| Ag | +1 (AgNO3) |
| Au | +1 (AuCl); +3 (AuCl3, KAuCl4) |
| Zn | +2 (ZnO, ZnSO4) |
| Hg | +1 (Hg2Cl2); +2 (HgO, HgCl2) |
| Sn | +2 (SnO); +4 (SnO2, SnCl4) |
| Pb | +2 (PbO, PbSO4); +4 (PbO2) |
Для многих элементов характерно несколько значений с.о. (табл. 13.1), и, вычислив его с.о., можно предвидеть окислительно-восстановительные свойства: элемент в наибольшей отрицательной степени окисления может только отдавать электроны (окисляться) и быть восстановителем, в наибольшей положительной с.о. – только принимать электроны (восстанавливаться) и быть окислителем, в промежуточных степенях окисления – и окисляться
|
|
|
|
|
Дата добавления: 2014-01-06; Просмотров: 1726; Нарушение авторских прав?; Мы поможем в написании вашей работы!