КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Потенциометрическое титрование в неводных средах
|
|
|
|
Потенциометрическое титрование можно проводить в неводных или смешанных растворителях. Необходимость применения неводных растворителей при титровании может возникнуть по различным причинам, из которых наиболее важными являются сила протолитов и растворимость вещества. Подбирая соответствующий растворитель, можно найти содержание компонентов, которые в водном растворе раздельно не титруются, провести анализ веществ, нерастворимых или разлагающихся в воде.
В подходящей неводной среде можно получить хорошие результаты при титровании смеси кислот, константы диссоциации которых в воде различаются менее чем на два порядка. Например, при потенциометрическом титровании в трет -бутанольном растворе смеси H2SO4 и НС1 раствором гидроксида тетрабутиламмония в изопропаноле на кривой титрования наблюдаются два скачка: первый соответствует титрованию НС1 и половины количества H2S04, а второй - нейтрализации HSO4- до SO42-.
Предел обнаружения F- при титриметрическом определении в смеси предельный спирт - вода снижается на 2 - 3 порядка. Уменьшение растворимости AgCl за счет введения в водный раствор 70% метанола дает возможность определять 2∙10-6 моль/л Сl- с хлорсеребряным-электродом с погрешностью около 2 %.
Наибольший интерес представляют методы кислотно-основного титрования неводных растворов органических веществ, анализ которых в водных растворах невозможен. В связи с этим рассмотрим известные представления о кислотах и основаниях.
Классическая теория кислот и оснований (С. Аррениус) основана на представлении об электролитической диссоциации веществ в водных растворах. Кислотами считаются вещества, при диссоциации которых образуются ионы водорода Н+; основаниями – вещества, диссоцирующие с образованием гидроксид-ионов ОН-. Подразделение на кислоты и основания, таким образом, основано на поведении веществ в водных растворах.
Согласно протолитической теории кислот и оснований Бренстеда и Лоури к кислотам относят вещества, которые способны отщеплять протоны. Вещества, способные присоединять протоны, называют основаниями. Такое деление веществ на кислоты и основания не связано с обязательным применением какого-либо растворителя. Протолитические взаимодействия могут протекать также между газообразными веществами. Однако даже при рассмотрении явлений в водных растворах протолитическая теория имеет существенные преимущества по сравнению с классической теорией. Это — общность описания кислотно-основных взаимодействий, в результате чего отпадает необходимость раздельного рассмотрения диссоциации и гидролиза.
Согласно протолитической теории кислоты и основания определяются соответственно как доноры или акцепторы протонов. Кислоты (доноры протонов) обычно обозначают буквой А, основания (акцепторы протонов) — буквой В (англ. base — основание). Пользуясь такими обозначениями, сказанное можно изобразить так:
А ↔ H+ + В (2.53)
Ради простоты заряды элементарных объектов кислоты и основания здесь не обозначены.
Процесс (2.53) обратим, поэтому всегда имеем дело с кислотно-основной или протолитической парой веществ. Каждый отдельный компонент протолитической пары (кислота, основание) называют протолитом. Кислота и основание одной и той же протолитической пары представляют собой сопряженные протолиты.
Приведем несколько примеров протолитических пар:
кислота основание
НС1 ↔ Н+ + С1-
СН3СООН ↔ Н+ + СНзСОО-
Н2СO3 ↔ Н+ + HCO3 -
НСО3 - ↔ Н+ + СО32-
Н3O+ ↔ Н+ + Н2O
H2O ↔ H+ + OH-
NH4+ ↔ Н+ + NH3
[Zn(OH2) n ]2+ ↔ Н+ + [Zn(OH) (OH2) n−1 ]+
[Zn(OH) (OH2) n-1 ]+ ↔ H+ + [Zn(OH)2 (OH2) n−2]
[Zn(OH)2 (OH2) n−2] ↔ H+ + [Zn(OH)3 (OH2) n−3]−
Из этих примеров видно, что протонодонорной способностью могут обладать как нейтральные молекулы, так и ионы. Поэтому кислоты подразделяют на молекулярные (НС1, HNO3, СН3СООН, Н2СO3, Н2O), катионные (H3O+, NH4+) и анионные (НСО3−). Аналогично основания делятся на молекулярные (NH3), катионные [Zn(OH)(OH2)n.1]+ и анионные (CI−, NO3−, СН3СОО−, НСО3−, ОН−). Некоторые протолиты (Н2O, НСО3−) обладают как протонодонорными, так и протоноакцепторными свойствами. Такие протолиты называют амфипротонными.
Приведенные примеры позволяют также судить о том, как с точки зрения протолитической теории рассматриваются классы веществ (кислоты, основания, соли), созданные на основании представлений классической теории. Вещества, которые классическая теория относит к кислотам, согласно протолитической теории представляют собой молекулярные кислоты, например НСl, СНзСООН. Основания классической теории, например NaOH, Ва(ОН)2, А1(ОН)з, Fe(OH)3 и т. п., с точки зрения протолитической теории состоят из двух протолитов. Гидратированный ион металла представляет собой кислоту, а гидроксид-ионы — основание. При этом основание значительно сильнее кислоты и реакция раствора определяется практически только присутствующим в нем основанием.
Соли с точки зрения протолитической теории тоже состоят из двух протолитов — из катионных кислот и анионных оснований. Так, например, в случае хлорида алюминия катионная кислота — гидратированные ионы алюминия, анионное основание — хлорид-ионы. В случае ацетата аммония катионная кислота — ионы аммония, анионное основание — ацетат-ионы и т. п. В водных растворах как катионные кислоты, так и анионные основания в большей или меньшей мере протолитически взаимодействуют с молекулами воды.
Силу кислотных и основных свойств отдельных компонентов протолитической пары теоретически можно оценивать с помощью констант соответствующих равновесий. Термодинамическая константа равновесия (2.53) служит для оценки силы кислоты, и ее называют константой кислоты:
КaA = (Н+)(В)/(А), (2.54)
здесь и далее круглыми скобками обозначены активности компонентов.
Сила основания оценивается константой обратного равновесия, называемой константой основания:
КaB=(А)/[(Н+)(В)]. (2.55)
Константа кислоты и константа основания сопряженных протолитов взаимосвязаны:
КaA = 1/КaB. (2.56)
Экспериментальным путем значения этих констант определить нельзя, поэтому ими можно пользоваться только при теоретических выкладках.
Причина этого заключается в том, что ни в одном растворителе концентрация свободных протонов не может достичь величины, при которой можно было бы определить их активность (Н+). После отщепления от одних протолитов протоны сразу же присоединяются к другим протолитам. Это и понятно, если учесть размеры протона (~10-15 м): он несравненно меньше атомов и молекул (размеры порядка 10-10 м). Поэтому у протона так сильно выражено стремление взаимодействовать с электронными орбиталями атомов и молекул. В результате всего этого в растворах свободных протонов практически нет.
Из сказанного вытекает, что для отщепления протона от кислоты A1 в системе должно присутствовать основание В2, которое этот протон принимает. Следовательно, химическое взаимодействие между протолитами, называемое протолитической реакцией, возможно только при наличии протолитов двух протолитических пар:
А1 = H+ + В1
B2 + H+ = A2
---------------------
A1 + B2 = B1 + A2 (2.57)
Например: СН3СООН + NH3 = СН3СОО− + NH4+.
Протолитическая реакция, как видно, протекает в результате обмена протоном между кислотой одной протолитической пары и основанием другой пары.
Условия протолитического равновесия (2.57) определяются термодинамической константой (Ka) этого равновесия:
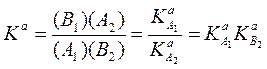 (2.58)
(2.58)
Важно отметить, что растворение протолитов в растворителях, молекулы которых способны к обмену протонами, является типичным кислотно-основным взаимодействием.
Приведем ряд примеров:
СH3COOH + H2O = CH3COO− + H3O+
H2O + NH3 = OH− + NH4+
HClO4 + СH3COOH = ClO4− + СH3COOH2+
Органические растворители, применяемые в потенциометрическом титровании в соответствии с донорно-акцепторными свойствами по отношению к протону можно разделить на апротонные (непротолитические), не участвующие в протолитических реакциях, и протонные (протолитические).
Апротонные растворители − это химические соединения (бензол, гексан, хлороформ, четыреххлористый углерод и т. п.), молекулы которых практически не способны ни отдавать, ни присоединять протоны. Эти растворители не вступают в протолитическое взаимодействие с растворенным веществом, а кислотно-основное равновесие в их средах осуществляется без участия растворителя.
В апротонных растворителях проявляется собственная кислотность или основность соединений. Поскольку апротонные растворители имеют низкие значения диэлектрической проницаемости, то кислоты, основания и соли в них заметно не диссоциируют. В этих растворителях преобладают процессы ассоциации, сопровождающиеся образованием ионных пар и продуктов присоединения молекул друг к другу.
Среди протонных растворителей различают кислотные или протогенные, способные отщеплять протон (НСООН, СНзСООН, С2Н5СООН и др.), основные или протофильные, способные связывать протон (жидкий аммиак, этилендиамин, пиридин и др.) и амфотерные, или амфипротные, обладающие кислотной и основной группами (этанол, пропанол, трет- бутанол, этиленгликоль и др.).
По способности изменять силу электролитов растворители делятся на дифференцирующие и нивелирующие. В дифференцирующих растворителях константы диссоциации кислот и оснований могут заметно различаться, даже если в воде они достаточно близки. Так, рK салициловой и пикриновой кислот в воде равны 2,97 и 0,8, а в ацетоне - соответственно 9,53 и 3,17. В нивелирующих растворителях сила кислот и оснований уравнивается. Каждый из растворителей проявляет в той или иной мере дифференцирующее или нивелирующее действие. Обычно протогенные растворители нивелируют силу оснований и дифференцируют силу кислот. Протофильные растворители, наоборот, нивелируют силу кислот и дифференцируют силу оснований. Например, анилин в жидком аммиаке - слабая кислота, а в уксусной кислоте - сильное основание.
Амфипротные растворители, к которым прежде всего относится самый распространенный растворитель — вода, являются более слабыми кислотами, чем протогенные растворители, и более слабыми основаниями, чем протофильные растворители.
При столкновении двух молекул амфипротонного растворителя HSolv одна из этих молекул проявляет протонодонорные свойства (кислота), а другая — протоноакцепторные свойства (основание), протекает протолитическая реакция, называемая реакцией автопротолиза (самоионизации), и устанавливается равновесие:
HSolv + HSolv = H2Solv+ + Solv- (2.59)
В 2 A1 A2 B1
Например, в случае воды:
H2O + H2О = Н3 O+ + ОН−
Ионы типа H2Solv+ называют ионами лиония (в частном случае Н3О+ — ионами гидроксония), ионы типа Solv− — ионами лиата (в частном случае ОН− — гидроксид-ионами).
Константа равновесия реакции автопротолиза (2.59) имеет следующее выражение:
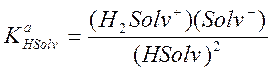 (2.60)
(2.60)
В чистом растворителе и в разбавленных растворах (HSolv) = 1, так как это соответствует термодинамическому стандартному состоянию этого вещества. Следовательно, константа автопротолиза (ионное произведение) растворителя равна произведению активностей ионов лиония и лиата:
KaHSolv = (H2Solv+) (Solv-), (2.61)
в случае воды: KaH2O = (Н3O+)(ОН−).
Равновесие автопротолиза в более или менее сильной мере смещено влево, и активности ионов лиония и лиата низки. Поэтому на практике удобно пользоваться отрицательными логарифмами
pK aHSolv = − lg K aHSolv; (2.62)
pH = − lg(H2Solv+); (2.63)
pSolv = − lg(Solv-). (2.64)
Уравнение (61) тогда принимает вид
pK aHSolv = pH + pSolv; (2.65)
для воды: рK aH2O = рН + рОН.
Нейтральность среды определяется равенством активностей ионов лиония и ионов лиата:
(H2Solv+) = (Solv−); рН = pSolv; рН = 1/2 рK aSolv.
в случае воды: (Н30+) = (ОН−); рН = рОН; рН = 1/2 рK aH2O.
Если (H2Solv+) > (Solv-); рН < pSolv; среда кислая:
в водных растворах: (HsO+) > (ОН-); рН < рОН.
Если (H2Solv+) < (Solv-); рН > pSolv; среда основная (щелочная)
в водных растворах: (Н3О+) < (ОН−); рН > рОН.
Степень кислотности или основности среды количественно можно оценить с помощью численных значений активностей ионов лиония и лиата. На практике для этой цели удобнее пользоваться показателями рН и pSolv [см. формулы (2.62) — (2.65)]. При рН = 0 имеем pSolv = рK aSolv, при pSolv = 0 рН = рK aSolv
Эти значения представляют собой естественные границы шкалы кислотности растворов. Следовательно, для каждого растворителя существует своя собственная нормальная шкала кислотности, которая простирается от рН = 0 до рН = рK aSolv.
Для иллюстрации сказанного в табл. 3 приведены данные по автопротолизу ряда амфипротонных растворителей. При этом следует отметить, что численные значения констант автопротолиза зависят от температуры, например, в случае воды:
| Температура, 0С pKH2O | 0 22 14,00 14,92 | 13,26 | 12,60 | 12,26 |
Таблица 3. Автопротолиз некоторых амфипротонных растворителей
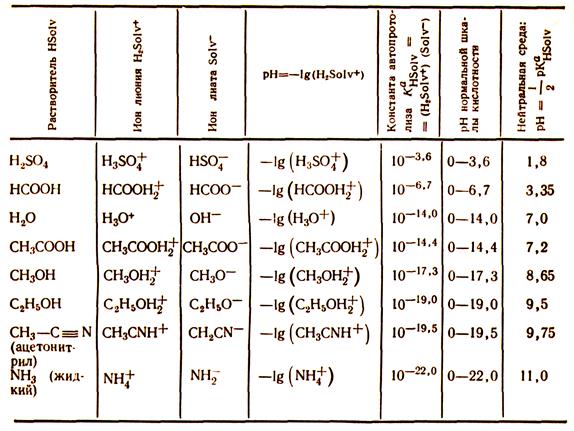
Кислоты и основания при растворении в амфипротонных растворителях вступают в протолитическое взаимодействие с растворителем. Вследствие этого изменяется активность (концентрация) ионов лиония и рН раствора в большей или меньшей степени отличается от рН нейтральной среды. Кроме того, раствор имеет определенные буферные свойства. При рассмотрении всех этих вопросов целесообразно протолиты подразделить на сильные и слабые.
Константа протолитической пары. Если растворенный протолит по отношению к молекулам растворителя проявляет кислые свойства, устанавливается следующее равновесие:
А + HSolv = H2SoIv+ + В (2.66)
в водных растворах: А + Н2O = Н3O+ + В
например: HCl + Н2O = Н3O+ + Сl−
CH3COOH + H2O = H3O+ + CH3COO−
NH4+ + H2О = H3O+ + NH3
Константу этого равновесия называют константой кислоты протолитической пары «А, В» в растворителе HSolv. Она связана с константой кислоты протолитической пары К aA, а также с константой кислоты KaH2Solv + протолитической пары «H2Solv+, HSolv» и с константой основания K a HSolv протолитической пары «H2Solv+, HSolv»:
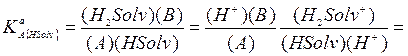
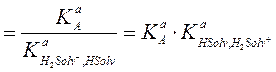 (2.67)
(2.67)
где 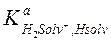 − константа равновесия процесса H2Solv+ = HSolv + H+,
− константа равновесия процесса H2Solv+ = HSolv + H+, 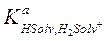 − константа основания (или основности) растворителя, т. е. константа равновесия процесса HSolv + H+ = H2Solv+, характеризующего протоноакцепторные свойства растворителя.
− константа основания (или основности) растворителя, т. е. константа равновесия процесса HSolv + H+ = H2Solv+, характеризующего протоноакцепторные свойства растворителя.
Как видно, значение константы КaA{HSolv} зависит как от кислотных свойств растворенного протолита (КaА), так и от основных свойств растворителя (Ka HSolv,H2Solv+). Поэтому один и тот же протолит оказывается тем более сильной кислотой, чем более сильными основными свойствами обладает растворитель.
Пример 1. В водных растворах ускусная кислота принадлежит к слабым кислотам и равновесие
СН3СООН + Н2О = СН3СОО- + Н3O+
смещено влево. Если воду заменить жидким аммиаком, основные свойства которого выражены гораздо сильнее, чем у воды, уксусная кислота становится сильной кислотой и равновесие
СН3СООН + NH3 → СН3СОО- + NH+
смещено вправо.
Пример 2. В водных растворах НСl является сильной кислотой и равновесие
HCI + Н2O → Н3O+ + CI-
смещено вправо. Если воду заменить безводной уксусной кислотой, у которой основные свойства выражены гораздо слабее, НС1 становится слабой кислотой и равновесие
НС1 + СН3СООН = СН3СООН2+ + С1-
смещено влево.
Если растворенный протолит по отношению к молекулам растворителя проявляет основные свойства, устанавливается равновесие:
В + HSolv = A + Solv- (2.68)
в водных растворах: В + H2O = А + OH−
например: NH3 + H2O = NH4+ + ОН−
Константу этого равновесия называют константой основания протолитической пары «А, В» в растворителе HSolv. Она связана с константой основания К а В протолитической пары «А, В» и с константой кислоты К a HSolv,Solv- протолитической пары «HSolv, Solv−»:
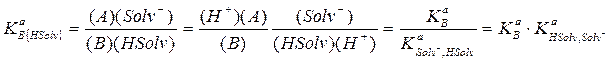 (2.69)
(2.69)
где 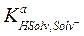 − константа кислоты растворителя, т.е. константа равновесия процесса HSolv = Solv- + H+, характеризующего протонодонорные свойства растворителя,
− константа кислоты растворителя, т.е. константа равновесия процесса HSolv = Solv- + H+, характеризующего протонодонорные свойства растворителя, 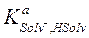 −константа равновесия процесса Solv- + H+ = HSolv.
−константа равновесия процесса Solv- + H+ = HSolv.
Следовательно, значение KaB{HSolv} зависит как от основных (протоноакцепторных) свойств растворенного протолита, так и от кислотных (протонодонорных) свойств растворителя. Чем сильнее выражены кислотные свойства растворителя, тем более сильным основанием оказывается данный протолит.
Между константами КaA{HSolv} и KaB{HSolv} одной и той же протолитической пары в одном и том же растворителе существует простая зависимость. Если перемножить уравнения (2.67) и (2.69), получим
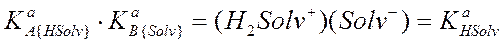 , (2.70)
, (2.70)
т.е. произведение констант кислоты и основания сопряженной пары в одном и том же растворителе равно константе автопротолиза этого растворителя. Это позволяет силу кислоты или основания в амфипротонном растворителе однозначно оценить с помощью одной из этих констант. На практике для этой цели обычно пользуются константой кислоты КaA{HSolv} и называют ее константой протолитической пары. Логарифмируя уравнение (70), получим
pКaA{HSolv} + pKaB{HSolv} = pKaHSolv. (2.71)
Следует отметить, что для водных растворов численные значения константы КaA{HSolv} совпадают с численными значениями константы диссоциации кислоты классической теории. В этом легко убедиться, сравнивая выражения этих констант. Например, для уксусной кислоты имеем
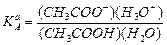 . Принимая для разбавленных растворов (H2O)=1 и (H3O+)=(H+), можно записать
. Принимая для разбавленных растворов (H2O)=1 и (H3O+)=(H+), можно записать 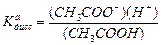
В случае оснований аналогично константа КaВ{HSolv} численно равна константе диссоциации основания классической теории. Благодаря таким взаимосвязям удобно пользоваться справочной литературой, составленной на основе представлений классической теории.
В табл. 3 приведены численные значения рK aA и рK aB для некоторых протолитических пар в водных, этанольных растворах, а также в растворах в безводной уксусной кислоте. Из таблицы видно, что принципиального различия между молекулярными и катионными (анионными) кислотами нет. То же характерно и для оснований.
Сопоставление данных табл. 4 для разных растворов показывает, как сильно могут изменяться кислотно-основные свойства протолитов при изменении амфипротонных растворителей.
Сила кислоты и основания одной и той же протолитической пары в соответствии с уравнением (2.70) взаимосвязана. Чем сильнее кислота, тем слабее сопряженное с ней основание, и наоборот. Если для оценки силы протолитов пользоваться значениями рK a А, можно сказать, что с увеличением этих значений уменьшается сила кислот и увеличивается сила оснований.
Таблица 4. Значения pKaA и pKaB для некоторых протолитических пар
| pK aA | Кислота А Основание В | pK aB |
| В водных растворах | ||
| ~ − 9 | НСlO4 + Н2O ↔ Н3O+ + СlO4- | ~23 |
| ~ − 8 | HI + Н2O ↔ Н3O+ + I- | ~22 |
| ~ − 7 | НС1 + Н2O ↔ Н3O+ + Сl- | ~21 |
| ~ − 3 | H2SO4 + H2O ↔ H3O+ + HSO4- | ~17 |
| − 1,4 | HNO3 + H2O ↔ H3O+ + NO3- | 15,4 |
| 0,0 | Н3O+ + Н2O ↔ Н2O + Н3O+ | 14,0 |
| 2,1 | Н3РO4 + Н2O ↔ Н3O+ + Н2РО4- | 11,9 |
| 3,2 | HF + Н2O ↔ Н3О+ + F- | 10,8 |
| 3,3 | HNO2 + Н2O ↔ Н3O+ + NO2- | 10,7 |
| 3,8 | HCOOH + H2O ↔ Н3O+ + HCOO- | 10,2 |
| 4,8 | CH3COOH+ H2O ↔ Н3O+ + CH3COO- | 9,2 |
| 6,4 | Н2СO3 + Н2O ↔ Н3O+ + НСО3- | 7,6 |
| 7,0 | H2S + Н2O ↔ Н3O+ + HS- | 7,0 |
| 7,2 | H2PO4-+ Н2O ↔ Н3O+ + HPO42- | 6,8 |
| 8,5 | [Cd(OH2)n]2+ + H2O ↔ Н3O+ + [Cd(OH) (OH2)n-1]+ | 5,5 |
| 9,1 | H3BO3 + Н2O ↔ Н3O+ + Н2ВО3- | 4,9 |
| 9,2 | NH4+ + Н2O ↔ Н3O+ + NH3 | 4,8 |
| 9,3 | HCN + Н2O ↔ Н3O+ + CN- | 4,7 |
| 9,4 | H4SiO4 + H2O ↔ Н3O+ + H3SiO4- | 4,6 |
| 9,9 | С6Н5ОН + Н2O ↔ Н3O+ + С6Н5O- | 4,1 |
| 10,3 | НСО3- + Н2O ↔ Н3O+ + СО32- | 3,7 |
| 12,4 | HPO42- + Н2O ↔ Н3O+ + PO43- | 1,6 |
| 12,6 | [Са(ОН2)n]2+ + Н2O ↔ Н3O+ + [Са(ОН) (ОH2) п -1]+ | 1,4 |
| 13,8 | HS- + Н2O ↔ Н3O+ + S2- | 0,2 |
| 14,0 | Н2O + Н2O ↔ Н3O+ + ОН- | 0,0 |
| NH3 + Н2O ↔ Н3O+ + NH2- | − 9 | |
| OH-+ Н2O ↔ Н3O+ + O2- | − 10 | |
| В этанольных растворах | ||
| 3,6 | HN03 + С2Н5ОН ↔ С2Н5ОН2+ + NO3- | 15,4 |
| 9,2 | НСООН + С2Н5ОН ↔ С2Н6ОН2+ + НСОО- | 9,8 |
| 10,3 | СН3СООН + С2Н5ОН ↔ C2H5OH2+ + СН3СОО+ | 8,7 |
| B безводной уксусной кислоте | ||
| 2,9 | НС104 + СН3СООН ↔ CH3COOH2+ + СlO4- | 11,5 |
| 4,1 | H2S04 + СН3СООН ↔ СН3СООН2+ + HSO4- | 10,3 |
| 5,0 | НС1 + СН3СООН ↔ CH3COOH2+ + Cl- | 9,4 |
| 9,4 | HN03 + CH3COOH ↔ CH3COOH2+ + NO3- | 5,0 |
протолитических пар
Протолиты подразделяют на сильные и слабые по смещениям равновесий в их растворах. Сильными называют протолиты, при растворении которых равновесия (2.66) или (2.68) смещены вправо. В случае кислот это имеет место при pKaA < О (KaA > 1), для оснований — при рK aA > pKaHSolv (pKaB < 0; КaB > 1). Таким образом, в водных растворах сильными кислотами считаются те, для протолитических пар которых рКaA < 0, а сильными основаниями — те, для протолитических пар которых рКaB > 14 (табл. 3). При растворении сильных кислот они в результате взаимодействия с молекулами растворителя практически полностью превращаются в ионы лиония. Другими словами, их сила в амфипротонном растворителе сводится к кислотной силе ионов лиония. Последние представляют собой самую сильную кислоту, существующую в данном амфипротонном растворителе.
Аналогичные рассуждения можно отнести и к сильным основаниям. При растворении их сила сводится к силе ионов лиата, которые представляют собой самые сильные основания в данном амфипротонном растворителе.
Следовательно, в водных растворах самая сильная кислота — ионы гидроксония и самое сильное основание — гидроксид-ионы.
Слабыми называют протолиты, для протолитических пар которых действительно неравенство 0 < pKaA < pK aHSolv (в случае водных растворов 0 < рКaA < 14). При растворении этих протолитов равновесия (2.66) и (2.68) смещены влево, и они превращаются в ионы лиония (лиата) только частично.
Подразделение на сильные и слабые протолиты, конечно, условно. Однако оно весьма полезно при решении ряда вопросов, в том числе при выводе приближенных формул для вычисления рН в растворах кислот и оснований. В таких формулах, как было показано выше, ввиду их приближенного характера целесообразно вместо активностей ионов пользоваться концентрациями. Правильное применение этих формул позволяет достаточно точно оценить рН в растворах многих протолитов.
При точном вычислении рН растворов сильных протолитов необходимо учитывать как те ионы лиония (лиата), которые образуются в результате взаимодействия протолита с молекулами растворителя, так и ионы лиония (лиата), которые возникают в результате автопротолиза. Однако в случае концентраций протолитов, применяемых обычно в аналитической химии, с влиянием автопротолиза растворителя можно не считаться. Только при очень низких концентрациях растворенных протолитов оба источника дают сравнимые количества ионов лиония (лиата). Так, например, для водных растворов сильных протолитов автопротолиз необходимо принимать во внимание только тогда, когда концентрация протолита ниже 10 -6 моль/л.
Согласно уравнениям (2.66) и (2.68) протолиз протекает в растворителях, имеющих высокие значения диэлектрической постоянной ε. При уменьшении значения этой постоянной увеличиваются силы электростатического взаимодействия между ионами и в растворах могут присутствовать в значительных количествах совокупности противоположно заряженных ионов. Такие совокупности называют ионными ассоциатами (ионными парами). Они возникают только вследствие электростатического притяжения и этим отличаются от молекул.
Если в растворе существуют ионные ассоциаты, следует различать две стадии взаимодействия протолитов с растворителем: перенос протонов и диссоциацию ионных ассоциатов. Например, в случае кислот вместо одного равновесия (2.66) необходимо рассматривать следующие равновесия:
перенос протонов: А + HSolv = [В; H2Solv+]
диссоциацию: [В; H2Solv+] = В + H2Solv+
Значение диэлектрической постоянной воды настолько велико (ε = 78,5), что равновесие диссоциации практически полностью смещено вправо и с присутствием ионных ассоциатов в водных растворах можно не считаться. В растворителях с низкими значениями диэлектрической постоянной образование ионных ассоциатов приводит к уменьшению силы кислоты (основания), так как уменьшаются количества возникающих ионов лиония (лиата). Например, за счет образования ионных ассоциатов можно объяснить уменьшение силы уксусной кислоты в этаноле (ε =24,2; рKaA = 10,3) по сравнению с силой этого протолита в воде (ε = 78,5; рKaА = 4,8).
На основании рассмотренных основных положений протолитической теории кислот и полученных соотношений можно сформулировать несколько практически важных выводов, которые позволяют правильно выбрать неводный растворитель, чтобы осуществить потенциометрическое титрование в оптимальных условиях.
1. Титрование очень слабой кислоты, согласно уравнения (2.67), следует проводить в полярном растворителе с высокой основностью. Наоборот, для усиления основных свойств слабых оснований (2.68) применяются кислые растворители, также по возможности с высокой диэлектрической постоянной. Так, азотистые органические основания (очень слабые основания в воде) взаимодействуют с кислотными растворителями, и поэтому их можно титровать как сильные основания, например, в безводной уксусной кислоте.
2. Если по каким-либо причинам замена растворителя невозможна, то при анализе очень слабого протолита в среде выбранного растворителя можно применять метод обратного титрования. Например, растворенный в воде фенол очень слабая кислота (КА {H2O}≈10-10) и не может быть определен титрованием сильным основанием. Сопряженное фенолу основание − фенолят-ион (C6H5O -), будет достаточно сильным основанием в воде. Согласно уравнению (2.70), значение КВ{H2O}= КН2О/КА{H2O}≈10-14/10-10≈10-4, поэтому фенолят можно оттитровать кислотой. Для проведения обратного титрования в анализируемый раствор фенола добавляют в избытке известное количество сильного основания (NaOH), затем титруют раствор сильной кислотой (HCl). На кривой титрования будет наблюдаться два скачка, первый из которых отвечает окончанию титрования избытка сильного основания, а второй – концу титрования слабого основания (фенолята).
3. При анализе смесей протолитов важно, чтобы растворитель был высоко дифференцирующим. Дифференцирующее действие растворителей зависит ряда факторов, а именно от кислотно-основных свойств, диэлектрической проницаемости, способности к образованию водородных связей и сольватирующей способности.
Дифференцирующее действие растворителей в первую очередь зависит от их кислотно-основных свойств. Например, в протогенных растворителях происходит дифференцирование силы кислот, т. е. большое количество веществ, которые в воде проявляют кислотные свойства, в кислых растворителях уже не являются кислотами. В среде муравьиной и уксусной кислот проявляют кислые свойства только минеральные кислоты, в то время как карбоновые кислоты таких свойств не проявляют.
По отношению к основаниям увеличение кислых свойств растворителя приводит к нивелированию силы оснований. В среде муравьиной кислоты почти все основания нивелированы по силе, а в среде уксусной кислоты основания, рКВ которых в воде более 10, также нивелированы.
Основные растворители, наоборот, нивелируют силу кислот и дифференцируют силу оснований. Органические соединения, проявляющие в некоторых растворителях основные свойства, не проявляют основных свойств в основных растворителях, сильные же в воде основания оказываются в них дифференцированными по силе.
4. Растворители амфипротонного характера обладают более высоким дифференцирующим действием как в отношении кислот, так оснований. Чем меньше константа автопротолиза, т.е. чем больше нормальная шкала рН растворителя, тем больше наблюдаемые при титровании в среде такого растворителя скачки титрования и тем больше возможность дифференцированного титрования смесей электролитов. Однако константы автопротолиза определены для немногих растворителей, в число которых не вошли некоторые растворители, получившие наибольшее практическое применение в аналитической химии (например, кетоны, нитропроизводные углеводородов, смешанные растворители). Поэтому большой практический интерес представляет определение относительной шкалы кислотности органических растворителей путем титрования в их среде наиболее сильных кислот и оснований, например хлорной кислоты и гидроокиси тетраариламмония. Указанные электролиты обычно используются в качестве наиболее сильных кислых или основных титрантов при определении оснований и кислот в неводных растворах.
Относительную шкалу кислотности данного растворителя выражают числом милливольт, получаемым путем вычитания потенциала полунейтрализации гидроокиси тетраэтиламмония из потенциала полунейтрализации хлорной кислоты как электролитов, которые занимают крайние положения на шкале кислотности:
ESolv=E1/2(A) – E1/2(B) (2.72)
где ESolv − относительная шкала кислотности растворителя, мB; E1/2(A) − − потенциал полунейтрализации НС1O4; E1/2(B) − потенциал полунейтрализации (C2H5)4NOH.
Значения потенциалов полунейтрализации кислот и оснований зависят от многих факторов, поэтому определение относительной шкалы кислотности каждого растворителя проводилось с одной и той же системой титрантов: хлорная кислота — гидроокись тетраэтиламмония и одной и той же системой электродов: стеклянный— насыщенный каломельный. Идеальным случаем явился бы тот, при котором растворы хлорной кислоты и гидроокиси тетраэтиламмония приготовлялись бы в среде исследуемого растворителя. Однако, если это условие выполнимо почти для всех случаев в отношении хлорной кислоты, то раствор гидроокиси тетраэтиламмония в силу некоторых технических причин или в силу нерастворимости гидроокиси тетраалкиламмония в некоторых растворителях готовился в среде смешанного растворителя: бензол—метиловый спирт, находящего наиболее широкое применение при титриметрических определениях в аналитической химии неводных растворов.
Для определения потенциалов полунейтрализации хлорной кислоты или гидроокиси тетраэтиламмония приготавливали 0,1 М раствор НС104 в исследуемом растворителе и 0,1 М раствор (С2Н5)4NОН в смеси бензол — метиловый спирт (5: 1). На титрование брали 25 [мл 0,1 М раствора кислоты или основания и помещали в 25 мл исследуемого растворителя. Таким образом, кислотный предел относительной шкалы кислотности растворителя определялся как потенциал полунейтрализации приблизительно 0,01 М раствора хлорной кислоты в исследуемом растворителе при титровании 0,1 М раствором гидроокиси тетраэтиламмония. В момент полунейтрализации хлорной кислоты исследуемый растворитель содержал менее 5 объемн. % бензольно-метаноловой смеси. Основной предел относительной шкалы кислотности растворителя определялся как потенциал полунейтрализации приблизительно 0,01 М раствора гидроокиси тетраэтиламмония в исследуемом растворителе, содержащем менее 10% бензольно-метаноловой смеси.
Размеры относительной шкалы кислотности спиртов, гликолей, кетонов, ацетонитрила, питропроизводных углеводородов, диметилформамида, пиридина и смешанных растворителей даны в табл. 5.
Сопоставление шкал кислотности исследуемых растворителей позволяет сделать некоторые выводы. Спирты характеризуются относительно небольшой величиной шкалы кислотности, которая для ряда исследованных спиртов (метилового, этилового и пропилового) близка к шкале кислотности воды. С увеличением радикала спирта и уменьшением диэлектрической проницаемости шкала кислотности увеличивается, например, при переходе от пропилового к изопропиловому и от бутилового к трет-бутиловому. Гликоли также обладают небольшой шкалой кислотности.
Кетоны характеризуются относительно большой шкалой кислотности. Наличие фенильных радикалов в молекуле кетонов приводит к уменьшению шкалы кислотности ароматических кетонов по сравнению с алифатическими. Ацетонитрил, так же как и кетоны, обладает большой шкалой кислотности. Нитрометан и нитробензол, а также диметилформамид имеют достаточно большие размеры шкалы кислотности, но эти шкалы расположены в различных областях ESolv (в мВ), так как нитропроизводные углеводородов характеризуются слабокислыми свойствами, а диметилформамид — слабоосновными. Пиридин, отличающийся более ярко выраженными основными свойствами по сравнению с диметилформамидом, имеет гораздо меньшую шкалу кислотности.
Таблица 5. Относительная шкала кислотности растворителей
| Растворитель | Диэлектрическая проницаемость | pKSolv | ESolv, мВ |
| Вода Этиленгликоль Диэтиленгликоль | 80,0 37,7 − 32,6 24,3 20,1 18,3 17,1 17,7 15,8 10,9 13,9 14,7 5,8 13,3 10,3 34,8 35,9 37,5 20,7 18,5 18,3 17,4 27,0 12,3 11,0* 8,4* 12,0* 9,3* 8,6* 6,0* | 14,00 15,60 − 16,70 18,95 19,33 20,84 26,5 18,00 | |
| Метиловый спирт | 32,6 | 16,70 | |
| Этиловый спирт | 24,3 | 18,95 | |
| Пропиловый спирт | 20,1 | 19,33 | |
| Изопропиловый спирт | 18,3 | 20,84 | |
| Бутиловый спирт | 17,1 | − | |
| Изобутилсвый спирт | 17,7 | − | |
| Втор- Бутиловый спирт | 15,8 | − | |
| Трет- Бутиловый спирт | 10,9 | − | |
| Амиловый спирт | 13,9 | − | |
| Изоамиловый спирт | 14,7 | − | |
| mpem-Амиловый спирт | 5,8 | − | |
| Гексиловый спирт | 13,3 | − | |
| Октиловый спирт | 10,3 | − | |
| Нитробензол | 34,8 | − | |
| Нитрометан Ацетон............................. | 35,9 | − | |
| Ацетонитрил | 37,5 | 26,5 | |
| Ацетон | 20,7 | − | |
| Метилэтилкетон | 18,5 | − | |
| Метилбутилкетон | − | − | |
| Циклогексанон | 18,3 | − | |
| Ацетофенон | 17,4 | − | |
| Метилбензилкетон | − | − | |
| Диметилформамид | 27,0 | 18,0 | |
| Пиридин | 12,3 | − | |
| Хлорбензол—метиловый спирт (4:1) | 11,0* | − | |
| Бензол—метиловый спирт (4:1) | 8,4* | − | |
| Хлорбензол—ацетонитрил (4:1) | 12,0* | − | |
| Бензол—ацетонитрил (4:1) | 9,3* | − | |
| Хлорбензол—ацетон (4:1) | 8,6* | − | |
| Бензол—ацетон (4:1) | 6,0* | − |
------------------------------------------
* Рассчитано по аддитивности
Кетоны характеризуются относительно большой шкалой кислотности. Наличие фенильных радикалов в молекуле кетонов приводит к уменьшению шкалы кислотности ароматических кетонов по сравнению с алифатическими. Ацетонитрил, так же как и кетоны, обладает большой шкалой кислотности. Нитрометан и нитробензол, а также диметилформамид имеют достаточно большие размеры шкалы кислотности, но эти шкалы расположены в различных областях ESolv (в мВ), так как нитропроизводные углеводородов характеризуются слабокислыми свойствами, а диметилформамид — слабоосновными. Пиридин, отличающийся более ярко выраженными основными свойствами по сравнению с диметилформамидом, имеет гораздо меньшую шкалу кислотности.
Добавление апротонных растворителей, константа автопротолиза которых теоретически стремится к нулю, к протолитическим приводит к уменьшению KSolv смешанных растворителей. С другой стороны, апротонные растворители характеризуются низкими значениями диэлектрической проницаемости, поэтому НСlO4 и (C2H5)4NOH в среде смешанных растворителей диссоциированы меньше, чем в среде протолитических растворителей. Это приводит к тому, что в одних случаях смеси апротонных растворителей с протолитическими растворителями имеют относительную шкалу кислотности больше, чем у протолитических растворителей, а в других случаях — меньше. Так, добавление апротонного растворителя к спиртам, имеющим небольшие шкалы кислотности, приводит к увеличению, а добавление к кетонам и нитрилам, которые обладают большими шкалами кислотности, приводит к уменьшению относительной шкалы кислотности.
Из этих выводов вытекают очень важные в практическом отношении положения, определяющие выбор растворителя для данного конкретного случая титрования. Согласно этим положениям, для дифференцированного титрования смесей электролитов в качестве сред следует использовать такие растворители, как кетоны, ацетонитрил, нитрометан, нитробензол, диметилформамид и смеси бензола или хлороформа с кетонами или ацетонитрилом. В среде этих растворителей получаются наиболее резкие скачки титрования и дифференцированно титруются многокомпонентные смеси кислот или оснований. Добавление углеводородов к спиртам способствует увеличению их дифференцирующего действия.
5. Величина диэлектрической проницаемости растворителей также оказывает существенное влияние на их дифференцирующее действие. Проследить влияние диэлектрической проницаемости на дифференцирующие свойства растворителей особенно отчетливо можно, сравнивая растворители с близкими кислотно-основными свойствами. Например, протогенные растворители — муравьиная и уксусная кислоты — значительно различаются величинами диэлектрической проницаемости (муравьиная кислота − ε =57, уксусная кислота − ε =6). Это различие сказывается на увеличении дифференцирующих свойств уксусной кислоты по сравнению с муравьиной.
В уксусной кислоте минеральные кислоты дифференцированы по силе, а в среде муравьиной кислоты эти кислоты хорошо ионизированы. Даже очень слабые в воде основания (рКВ >10—14) в среде уксусной кислоты также дифференцированы по силе, в то время как в муравьиной кислоте большинство оснований имеет рКВ ~1. Исследование диссоциации кислот в монохлоруксусной (ε = 20), трихлоруксусной (ε = 4,5) и масляной (ε = 2,4) кислотах также показало, что дифференцирующее действие кислот возрастает  с уменьшением диэлектрической проницаемости.
с уменьшением диэлектрической проницаемости.
Влияние диэлектрической проницаемости на дифференцирующие свойства растворителей можно проследить и при рассмотрении диссоциации кислот в основных растворителях. Так, гидразин (ε=52) нивелирует силу кислот, а пиридин (ε=12,5) оказывает дифференцирующее действие. Сила кислот и соотношение в их силе в пиридине близки к силе и соотношению в уксусной кислоте, что подтверждает вывод о том, что влияние этих растворителей в значительной степени обусловлено их низкой диэлектрической проницаемостью.
Апротонные растворители являются растворителями с высокими дифференцирующими свойствами вследствие низкого значения диэлектрической проницаемости этих растворителей. Поэтому часто для дифференцирования силы протолитов используют смеси из амфипротонных и апротонных растворителей.
Рассматривая зависимость дифференцирующего действия растворителей одновременно от кислотно-основных свойств растворителя и величины диэлектрической проницаемости, можно сказать, что в одних случаях наибольшее значение имеют кислотно-основные свойства растворителя, а в других — величина диэлектрической проницаемости.
Например, высокие дифференцирующие свойства ацетонитрила обусловлены его малыми кислотно-основными свойствами, так как его диэлектрическая проницаемость относительно велика (ε = 37,5). То же можно сказать и о дифференцирующем действии формамида (ε =105) в отношении кислот, а также нитрометана (ε = 35,9) и нитробензола (ε = 34,8) в отношении оснований. С другой стороны, как было показано, дифференцирующее действие уксусной кислоты и пиридина, являющихся растворителями с ярко выраженными протогенными и протофильными свойствами, соответственно обусловлено низкими значениями диэлектрической проницаемости.
6. В значительной мере дифференцирующее действие растворителей зависит от сольватирующей способности и способности молекул растворителя образовывать водородные связи с молекулами электролита. Диссоциация электролита в растворе часто идет по схеме:
HA + n Solv ↔HA(Solv) n ↔ HSolv+cольв. + A- cольв.,
т. е. на ионы диссоциирует не сама кислота, а продукт ее присоединения к растворителю. Следовательно, характер и прочность водородных связей оказывают существенное влияние на диссоциацию продукта присоединения и, следовательно, на дифференцирующие свойства растворителей.
Наиболее резкое изменение в относительной силе протолитов можно ожидать при переходе от растворителей, которые могут быть акцепторами и донорами протонов (вода, спирты), к растворителям, у которых акцепторные свойства преобладают над донорными (кетоны, нитрилы, питросоединения).
Для каждого конкретного случая выбор растворителя имеет важное практическое значение, так как позволяет осуществить потенциометрическое титрование в оптимальных условиях. Однако этот выбор затруднен из-за ограниченности сведений о константах ионизации протолитов в неводных растворителях.
Простым, удобным и быстрым методом оценки влияния растворителей на силу кислот и оснований, также как и для оценки относительной шкалы кислотности растворителей, является метод определения относительной кислотности электролитов по потенциалам полунейтрализации. В момент, когда нейтрализовано 50% определяемой слабой кислоты или слабого основания, рН = рК. Следовательно, величина потенциала полунейтрализации определяется величиной константы ионизации титруемого электролита и может характеризовать его относительную силу в неводных растворах.
Абсолютные значения потенциалов полунейтрализации в неводных растворах нестабильны, поэтому измеряют потенциалы полунейтрализации по отношению к потенциалу полунейтрализации стандартного вещества. В качестве стандарта при определении потенциалов полунейтрализации кислот обычно используют бензойную кислоту, а при определении потенциалов полунейтрализации оснований — дифенилгуанидин. Разность Е(1/2) исследуемой кислоты или основания и стандартного вещества дает представление о возможности дифференцированного титрования смеси кислот (оснований), т.е. величина ΔE(1/2) может служить критерием их силы в неводных растворах
ΔЕ(1/2) = Е(1/2),x – E(1/2),ст., (2.73)
где ΔЕ(1/2),x, и ΔE(1/2),ст. − потенциалы полунейтрализации исследуемого и стандартного вещества. При определении ΔЕ(1/2) существенную роль играют сила растворенных кислот и оснований, а также природа растворителя и растворенного вещества. Чем больше величина ΔE(1/2), тем выше сила протолита и дифференциирующая способность растворителя. Различие потенциалов полунейтрализации в 200 - 300 мВ в большинстве случаев оказывается достаточным для осуществления избирательного титрования.
Было показано, что зависимость ΔЕ(1/2) в данном растворителе от значений показателей констант диссоциации кислот или оснований в воде выражается, как правило, прямой линией, но каждой природной группе кислот или оснований соответствует своя прямая.
О дифференцирующем действии неводных растворителей можно судить по зависимости потенциалов полунейтрализации от рК (Н2О). Дифференцирующее действие растворителя характеризуется наклоном прямой: чем круче идет прямая, тем более высоким дифференцирующим действием обладает растворитель в отношении данной группы электролитов. Так, например, наклон зависимости от pKA(H20) для аминов в воде, уксусной кислоте и ацетонитриле составляет соответственно 51, 59 и 100 мВ на единицу рКа (Н2О). Это показывает, что лучшими дифференцирующими свойствами среди этих трех растворителей обладает ацетонитрил, при титровании в среде которого смесей аминов можно получить наиболее резкие конечные точки титрования.
Для выяснения возможности дифференцированного определения смесей аминов и диаминов измерены ΔЕ(1/2) в среде спиртов, кетонов и смешанных растворителей. При этом для всех исследованных растворителей зависимость ΔЕ(1/2) от рКА (Н2О) оказалась линейной (рис. 2.14). Причем для аминов и диаминов получена общая линейная зависимость. Сравнение дифференцирующих свойств спиртов, кетонов и смешанных растворителей в отношении аминов и диаминов показало, что наклон прямой [в милливольтах на единицу рК (Н20)] составляет: для метилового спирта — 41, изопропилового спирта—42, ацетона —47, метилэтилкетона — 54, смеси хлороформ — метилэтилкетон (4:1)—67, смеси хлороформ—ацетонитрил (4:1) — 62. Следовательно, наиболее подходящей средой для дифференцированного титрования смесей аминов и диаминов являются смешанные растворители.
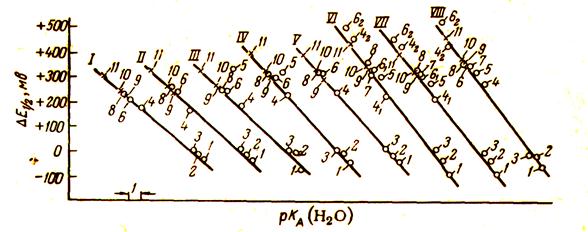
Рис. 2.14. Относительная сила аминов и диаминов в среде:
I —метилового спирта; II —изопропилового спирта; III —ацетона; IV— метилэтилкетона; V— хлороформа— изопропилового спирта (4: 1); VI— хлороформа—метилэтилкетона (4: 1); VII— бензола—
метилэтилкетона (4: 1); VIII —хлороформа—ацетонитрила (4: 1). 1 —пиперидин; 2—н -бутиламин; 3 —дифенилгуанидин; 4—п -фенилендиамин; 5 —акридин; 6—м -фенилендиамин; 7 —бензидин; 8— о-фенилендиамин; 9— м -толуилендиамин; 10—п -аминодифениламин; 11 —1,5- нафтилендиамин.
Аппаратура и методы титрования в неводных средах. Титрование в неводных растворах обычно осуществляют полумикрометодом. Для титрования используют установки (рис. 2.15) с автоматическими микробюретками. Емкости таких бюреток составляют 5, 2 или 1 мл с ценой деления 0,01 мл, емкость баллона для хранения титранта — 250, 200 или 100 мл соответственно.
Определенные трудности при проведении кислотно-основного титрования создает диоксид углерода, поглощаемый из воздуха. В диапазоне рН 6-8 поглощение СО2 вызывает дрейф показаний прибора, что затрудняет получение точного значения ЭДС. Для сведения к минимуму поглощения диоксида углерода через анализируемый раствор целесообразно пропускать газообразный азот.

Рис. 2.15. Схема установки для потенциометрического титрования:
1 —автоматическая полумикробюретка; 2—стакан для титрования; 3 —электроды; 4 —потенциометр; 5— магнитная мешалка; 6 —осушительные системы; 7 —барботер; 8 —промежуточная склянка; 9 —баллон с азотом; 10— кран.
Титрование осуществляют в изолированной от внешней среды ячейке 2 емкостью 50—100 мл, в которой сделаны отверстия для кончика бюретки (см. рис. 2.15), трубки, по которой подается сухой азот, и электродов. При титровании кончик бюретки должен находиться вблизи уровня жидкости или быть опущенным в титруемый раствор. Очень слабые кислоты титруют в токе сухого азота,
Для титрования соединений основного характера в неводных растворах в качестве титрантов используют в основном неводные растворы хлорной кислоты. Хлорная кислота является одной из самых сильных кислот в среде неводных растворителей, что и обуславливает ее широкое применение. В качестве титрантов применяются также алкил- или арилсульфоновые кислоты. В уксуснокислой среде фторсульфоновая кислота является более сильной по сравнению с хлорной, а стабильность ее растворов и простота приготовления позволяют считать ее одним из лучших титрантов. Иногда используются также растворы салициловой, пикриновой, трихлоруксусной, иодной, азотной и серной кислот.
Из неорганических оснований наиболее широко применяются метаноловые, этаноловые, изопропаноловые и смешанные стандартные растворы едкого кали и едкого натра. С успехом используются уксуснокислые растворы ацетатов щелочных металлов, которые по силе не уступают спиртовым растворам щелочей. Широко применяются спиртовые растворы алкоголятов щелочных, металлов: метилаты, этилаты, изопропилаты натрия, калия, лития в среде соответствующих спиртов и аминоэтилат натрия в среде этилендиамина.
Самыми сильными основными титран
|
|
|
|
|
Дата добавления: 2014-01-07; Просмотров: 4618; Нарушение авторских прав?; Мы поможем в написании вашей работы!