КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Ступінь дисоціації електроліта – це відношення числа молекул, що розпалися на іони, до загального числа молекул розчиненої речовини
|
|
|
|
Інакше кажучи,αі – доля молекул електроліта, що розпалися на іони:
α і = n/N.
За ступенем дисоціації електроліти умовно поділяють на сильні (αі > 30%) і слабі (αі < 3 %). У проміжку 3 % < α і < 30 % електроліти середньої сили.
На ступінь дисоціації впливає:
- температура,
- концентрація,
- додавання однойменних іонів.
Кількісно електролітичну дисоціацію як рівноважний оборотній процес можна охарактеризувати константою дисоціації (іонізації), що визначається законом діючих мас.
Закон діючих мас, точно говорячи, застосовується лише до оборотних реакцій, тобто до розчинів слабких електролітів. Якщо дисоціацію електроліта уявити як рівноважний процес:
Kt n An m ↔ nKt m+ + mAn n-.
Відповідно до закона діючих мас константу рівноваги запишемо так:
Kд = [Kt m+ ] n [An n- ] m / [Kt n An m ].
Це рівняння справедливе лише для розбавлених розчинів слабких електролітів. Чим більша константа дисоціації Кд, тим сильніший електроліт.
На відміну від ступеня дисоціації Кд залежить тільки від природи електроліта і температури, але не залежить від концентрації розчину! Таким чином, і константа дисоціації Кд, і ступінь електролітичної дисоціації αі – кількісні характеристики дисоціації. Зрозуміло, що між ними існує зв’язок.
Так для слабкого бінарного електроліта концентрація
[Kt + ] = [An - ] = с(Х)αі,
а концентрація недисоційованих молекул
[KtAn] = с (Х) – с (Х)*αі = = с (Х) (1 – αі).
Підставивши ці значення у вираз для константи дисоціації, одержимо:
Кд = с (Х)*αі*с (Х)*αі / с (Х)*(1 - αі) = αі2*с (Х)/ (1 - αі).
Це співвідношення називають законом розведення Оствальда (1888).
Для слабкого електроліта αі << 1, тоді величиною αі у знаменнику можна знехтувати і рівняння прийме вигляд:
Кд ≈ αі2*с (Х) або αі ≈ √Кд/с (Х).
Якщо замість 1/с (Х) підставити V(Х) = 1/с (Х), яке називається розведенням, рівняння набуде вигляду
αі ≈ √КдV (Х).
Відповідно закон Оствальда може бути сформульований так:
ступінь дисоціації слабкого електроліту зростає з розведенням розчину.
Сильні електроліти не підкоряються цьому закону.
Багатоосновні кислоти і багатокислотні основи дисоціюють ступінчасто. Дисоціація за ступенями характеризується ступінчастими константами. На прикладі фосфорної кислоти.
Сумарне рівняння. Сумарна константа дисоціації Ксум = К1К2К3. При цьому К1 > К2 > К3. Замість константи дисоціації зручніше користуватися її десятковим логарифмом, взятим з оберненим знаком: рКд = - lg Кд. Наприклад для оцтової кислоти Кд = 1,76*10-5, і відповідно рКд = 4,76.
ТЕОРІЯ РОЗЧИНІВ СИЛЬНИХ ЕЛЕКТРОЛІТІВ ДЕБАЯ І ХЮККЕЛЯ.
У водних розчинах сильні електроліти практично повністю дисоційовані.
У зв’язку з цим для оцінки концентраційних ефектів у розчинах сильних електролітів вводиться величина, яка називається активністю α:
Під активністю електроліта Х розуміють ефективну концентрацію, у відповідності до якої він приймає участь у різних процесах.
Активність зв’язана з істиною концентрацією розчиненої речовини співвідношенням:
α (Х) = f (Х)*с (Х),
де α (Х) – активність електроліта, моль/л; с (Х) – його концентрація, моль/л; f (Х) – коефіцієнт активності (величина безрозмірна).
Коефіцієнт активності f (Х) виражає відхилення розчину з концентрацією с (Х) від поведінки розчину при безкінечному розведенні, тобто за відсутності між іонних взаємодій.
ІОННА СИЛА РОЗЧИНУ.
В розведених розчинах природа іонів незначно впливає на значення коефіцієнтів активності, оскільки між іонні взаємодії визначаються лише зарядами іонів та їх концентрацією. При цьому кількісною характеристикою між іонних електростатичних взаємодій є іонна сила розчину І:
Іонною силою розчину називають величину, що вимірюється напівсумою добутку концентрацій усіх іонів, що знаходяться у розчині, на квадрат їх заряду.
І = ½ ∑(сіzі2),
де І – іонна сила розчину; сі – молярні концентрації іонів; zі – заряди іонів.
П. Дебай і Е. Хюккель в 1923 р. показали, що для розбавлених розчинів з іонною силою І≤ 0,01 коефіцієнти активності можна розрахувати за формулою:
lg fi = - 0,5 zi2√I,
де fi – коефіцієнт активності іона, а zi – його заряд.
Якщо користуватися значеннями активностей замість концентрацій, то закон діючих мас можна застосовувати до сильних електролітів і концентрованих розчинів слабких електролітів. Для таких розчинів константу рівноваги слід виражати через активності.
ДИСОЦІАЦІЯ ВОДИ.
Як уже було показано, вода поводе себе як амфоліт. Процес дисоціації води відповідно до теорії Бренстеда протікає за рівнянням
H2О + H2О ↔ Н3О+ + ОН-
Відбувається автоіонізація води.
Константа дисоціації води при 25 0С дорівнює:
Кд (Н2О) = а (Н+)* а (ОН-)/ а (Н2О) = 1,8*10-16 моль/л, де а (Н+), а (ОН-), а (Н2О) – активності іонів Н+, ОН- та води Н2О.
Ступінь дисоціації води дуже малий, тому активності гідроген- та гідроксид-іонів в чистій воді практично дорівнюють їх концентраціям. Оскільки вода присутня у великому надлишку, її концентрація може вважатися сталою й складає 55,6 моль/л (1000: 18 г/моль = 55,6 моль/л). Підставивши це значення до виразу для константи дисоціації, а замість активностей гідроген- та гідроксид-іонів їх концентрації, одержують новий вираз:
К (Н2О) = [Н+][ОН-] = 1*10-14 моль2/л2, або точніше К (Н2О) = а (Н+)* а (ОН-) = 1*10-14 моль2/л2. Константа К (Н2О) називається іонним добутком або константою автоіонізації (автопротолізу) води. Тобто в чистій воді або любому водному розчині при сталій температурі добуток концентрацій (активностей) гідроген- та гідроксид-іонів є величина стала, яка називається іонним добутком води.
Константа К (Н2О) залежить від температури: при підвищенні температури К (Н2О) зростає.
В чистій воді активності а (Н+) = а (ОН-) = 1*10-7 моль/л:
а (Н+) = а (ОН-) = 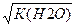 = √10-14 = 1*10-7
= √10-14 = 1*10-7
Якщо до чистої води додати стільки лугу, щоб концентрація гідроксид-іонів підвищилася, наприклад. до 10-4 моль/л, то концентрація [Н+] знизиться до 10-10 моль/л. Так що за законом діючих мас іонний добуток води залишиться рівним 1*10-14 моль2/л2.
Навпаки, якщо до чистої води додати стільки кислоти. Щоб концентрація іонів гідрогену підвищилася, наприклад, до 10-3 моль/л, то концентрація гідроксид-іонів знизиться до 10-11 моль/л, а іонний добуток води знову 1*10-14 моль2/л2.
Отже, знаючи концентрацію одного з цих іонів, завжди можна розрахувати концентрацію іншого іона. Як правило, для характеристики кислотності середовища використовують від’ємний десятковий логарифм активності (концентрації) гідроген-іонів, який називається водневим покажчиком рН середовища:
рН = -lg а (Н+)
або наближено рН = -lg а [Н+]
Нейтральне середовище рН = 7, кисле - рН < 7, лужне – рН >7.
Реакцію середовища можна характеризувати й гідроксид ним покажчиком: рОН = -lg а (ОН-)
або наближено рОН = -lg а [ОН-]
Тоді логарифмування виразу іонного добутку води дасть рН + рОН = 14.
В розчинах слабких кислот НА кислотно-основна рівновага ІІІ типу має вигляд
НА + Н2О ↔ Н3О+ + А- або НА ↔ Н+ + А-
Константа кислотної дисоціації К а дорівнює:
К а = [Н+][А-] / [НА].
У стані рівноваги [Н+]=[А-], тоді [НА] = (с (НА) - [Н+]). Підставивши це у вираз для константи, одержимо К а = [Н+]2 / (с (НА) - [Н+]). Якщо загальна концентрація кислоти с (НА) більша як 0,01, значенням [Н+] у знаменнику можна зневажити і рівняння набуде вигляду К а = [Н+]2 / с (НА).
Тоді
[Н+] = √ К а с (НА),
рН = ½ р К а – ½ lg с (НА),
де р К а = - lg К а.
Для розчинів слабих основ формули для розрахунку рН вивести самостійно!
За аналогією до К а – константою кислоти існує й К b – константа основи. Між К а та К b є певний зв’язок: К а К b = 10-14
рК а + рК b = 14.
БУФЕРНІ РОЗЧИНИ. ТИПИ БУФЕРНИХ СИСТЕМ.
Часто під час експериментальних досліджень у хімії, біології, медицині, у промисловому виробництві виникає потреба забезпечити сталість рН середовища, оскільки внаслідок перебігу хімічних реакцій можуть утворюватись або витрачатись іони Гідрогену. Щоб процес відбувався за сталого значення рН, у розчин вводять буферні системи, які підтримують рН середовища практично незмінним. Фізіологічні рідини організму характеризуються сталим значенням рН. Це досягається як за допомогою фізіологічних (за участю таких органів, як нирки, печінка, легені, кишки), так і фізико-хімімічних механізмів (завдяки дії буферних систем).
|
|
|
|
|
Дата добавления: 2014-01-07; Просмотров: 1296; Нарушение авторских прав?; Мы поможем в написании вашей работы!