КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Ионоселективные электроды
|
|
|
|
Ионоселективные электроды отличаются от всех рассмотренных ранее тем, что у них обе граничащие фазы - мембрана и раствор - обладают ионной проводимостью, и поэтому на их границе не происходит собственно электрохимическая реакция с переносом электронов. Процесс сводится здесь к обмену ионами между мембраной и раствором. Межфазную границу пересекают только ионы, заряд которых при этом не изменяется. При соответствующем подборе состава и структуры мембраны потенциал на межфазной границе будет зависеть от активности только одного какого-либо вида ионов. Такие электроды обладают, следовательно, селективностью и позволяют измерять активность отдельных ионов (конечно, с известными ограничениями, обусловленными термодинамической невозможностью определения активностей отдельных ионов).
Мембраны ионоселективных электродов могут быть твёрдыми и жидкими. К твёрдым мембранам относятся стеклянные, кристаллические и гетерогенные. К жидким - не смешивающиеся с водой органические растворители с низкой диэлектрической постоянной (хлорбензол, толуол и др.), в которых растворены соответствующие ионогены - диэфиры фосфорной кислоты, алифатические кислоты, амины и др.; кроме того, подобные же растворители, но с растворёнными в них органическими веществами с относительно крупными молекулами (например, краун-эфиры), связывающими ионы и переносящими их из водной среды в органическую.
Стеклянные электроды, обратимые по отношению к ионам водорода, были первыми ионоселективными электродами. Они изобретены в начале ХХ в. Кремером, Габером и Клеменсиевичем. Квантово-механический вариант теории стеклянного электрода предложил М.Дол (1934), а её термодинамический вариант, получивший наибольшее распространение и ставший основой последующего развития теории ионоселективных электродов - Б.П.Никольский (1936).
В стёклах известной подвижностью обладают лишь низкозарядные катионы, в первую очередь, ионы щелочных металлов, а силикатные, алюмосиликатные или иные оксиды образуют практически неподвижную сетку. При создании контакта между стеклянной мембраной и раствором начинается обмен ионами между граничащими фазами, например между ионами щелочного металла М+(*) в стекле и ионами водорода в растворе
Н+(*) + М+ = Н+ + М+(*)
(где знак * относится к мембране). Псевдоравновесное состояние, достигаемое в ходе обмена, характеризуется константой обмена
К = 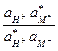 .
.
Предполагая, что в стекле данного сорта сумма активностей ионов металла и водорода постоянна и равна активности ионов металла в исходном стекле
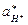 =
= 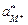 = а,
= а,
можно преобразовать вышеприведенное уравнение для К:
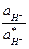 =
= 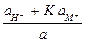 .
.
Потенциал стеклянного, как и любого другого ионоселективного электрода, нельзя измерить, просто сочетая его с электродом сравнения, подобно тому как это делалось с уже рассмотренными электродами, поскольку ионоселективные электроды не обладают электронной проводимостью. Можно, однако, измерить разность потенциалов между сторонами мембраны, включив её в электрохимическую систему с двумя одинаковыми электродами и приняв меры по устранению диффузионного потенциала
AgïAgClï0,1M NaClïМембранаïИсслед. р-рïê0,1M NaClïAgClïAg
В системе слева и справа находятся одинаковые хлорсеребряные электроды, разность потенциалов между которыми при соответствующей процедуре их приготовления пренебрежимо мала. Правая сторона мембраны контактирует с исследуемым раствором. Потенциал левой стороны мембраны, контактирующей с раствором внутреннего электрода сравнения, определяется аналогичным уравнением, но с иными значениями активностей Н+ и М+.
Состав раствора с левой стороны мембраны сохраняется неизменным, диффузионный потенциал в системе мал и тоже почти постоянен; их можно поэтому объединить в константу. В неё же можно включить небольшую разность, обусловленную некоторым различием в физических и химических свойствах двух сторон мембраны и называемую потенциалом асимметрии. В конце концов для потенциала ионоселективного стеклянного электрода можно получить следующее уравнение:
Ест = Еост + 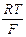 ln (
ln (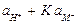 ). (1)
). (1)
В зависимости от значения К электрод будет селективным по отношению к ионам водорода (К << 1) или к ионам металла (К >> 1), либо вообще не будет обладать селективностью (К = 1). Стеклянные электроды, предназначенные для измерения рН, имеют К порядка 10–12 - 10–10, поэтому в кислых, нейтральных и даже в слабощелочных системах 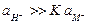 и уравнение упрощается до
и уравнение упрощается до
Ест = Еост + ln 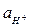 = Еост – 0,059pH, (2)
= Еост – 0,059pH, (2)
то есть в таких растворах потенциал стеклянного электрода зависит только от активности ионов водорода.
Если 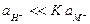 , то есть в щелочной области, уравнение также упрощается и принимает вид, где в величину Еост¢ входит слагаемое, содержащее константу обмена
, то есть в щелочной области, уравнение также упрощается и принимает вид, где в величину Еост¢ входит слагаемое, содержащее константу обмена
Ест = Еост¢ + ln 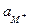 = Еост¢ + 0,059 lg . (3)
= Еост¢ + 0,059 lg . (3)
В щелочных растворах потенциал стеклянного электрода зависит от активности катиона щёлочи и, следовательно, его можно использовать в качестве индикаторного электрода для определения активности ионов соответствующего щелочного металла. Если источником катионов служит только раствор щёлочи, тогда =  , а так как
, а так как 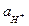 ×= KW, то для щелочной области растворов вместо (3) можно написать
×= KW, то для щелочной области растворов вместо (3) можно написать
Ест = Еост¢¢ – ln = Еост¢¢ + 0,059pH, (4)
откуда следует, что в кислых и щелочных растворах потенциал стеклянного электрода является функцией активности ионов водорода. Каждой области рН отвечает при этом своё значение стандартного потенциала Ео стеклянного электрода, а наклоны прямых Ео - рН в кислой и щелочной областях одинаковы по величине и обратны по знаку. Зависимость потенциала стеклянного электрода от рН выражается прямой с максимумом, положение которого зависит от сорта стекла и определяется его константой обмена. В действительности, однако, поведение ионоселективных электродов сложнее, чем то, которое легло в основу рассмотренной теории стеклянного электрода.
Уравнение (1) формально приложимо к любым ионоселективным электродам, но физический смысл коэффициента селективности в каждом конкретном случае оказывается иным, и иными будут факторы, определяющие эффективную работу электрода. Так, в случае твёрдых кристаллических электродов (галогениды серебра, сульфид серебра, трифторид лантана), применяемых для определения активностей ионов серебра, галогенид-ионов, сульфидов и фторидов, важное значение приобретает растворимость и комплексообразование соответствующих соединений. Это же относится и к гетерогенным мембранам на основе труднорастворимых соединений, распределённых в непроводящей связке, например, в силиконовом каучуке. В жидких мембранах присутствуют, в зависимости от природы ионизируемого агента, органофильные катионы либо анионы и обычные противоионы, способные обмениваться с катионами в водном растворе. Вид уравнения для потенциала жидкой ионоселективной мембраны зависит от степени диссоциации (ассоциации).
Лекция 59
Электролиз. Токи обмена. Поляризация электрода, перенапряжение. Концентрационная и электрохимическая поляризация. Напряжение разложения
Кинетика электрохимических процессов
Равновесные состояния процессов внутри электролитов (электролитическая диссоциация, гидролиз, сольватация и другие) и процессов на электродах (электрохимические реакции и характеризующие их обратимые электродные потенциалы) не зависят от времени, к ним применимы оба закона термодинамики. Поэтому соответствующие закономерности называются термодинамическими, а посвященный им раздел электрохимии – термодинамикой электрохимических процессов. Для электродных процессов равновесие характеризуется отсутствием электрического тока.
Процесс прохождения электрического тока конечной силы не является равновесным, и явления, связанные с прохождением тока, зависят от времени и от силы тока, величина которого может быть регулируема извне. Раздел электрохимии, рассматривающий неравновесные, главным образом стационарные процессы, протекающие на электродах во времени, называется кинетикой электрохимических (электродных) процессов или просто электрохимической кинетикой.
Электрический ток может протекать в результате замыкания электрохимического элемента, образуемого электродами и электролитом, или под влиянием приложенной к системе электроды – электролит внешней разности потенциалов. В последнем случае явления, происходящие на границах электрод – электролит, называются электролизом и состоят в выделении веществ (металлы, газы) из электролита на электроде, в растворении вещества электрода и в изменении состава электролита.
Электрохимическая кинетика основывается как на общих положениях химической кинетики, так и на частных закономерностях, характерных только для электрохимических процессов. Так, для электрохимии справедливы основной постулат химической кинетики, применимость понятия энергии активации для многих электрохимических процессов, положительное влияние температуры на скорость электролиза и т.п.
Достаточно отчетливо выражена и специфичность электрохимических процессов:
1. Электрохимическим путем можно проводить и такие реакции, которые химическим путем при обычной температуре не идут (например, реакция разложения воды при обычной температуре не идет, а электролизом вода легко разлагается). Самопроизвольные реакции всегда сопровождаются уменьшением свободной энергии; электрохимическим же путем можно проводить реакции, сопровождающиеся увеличением свободной энергии, то есть возможности электросинтеза шире, чем возможности обычного химического синтеза. Необходимая свободная энергия доставляется системе извне в виде энергии электрического тока.
2. Суммарную скорость электрохимического процесса можно не только легко определить по величине силы тока, протекающего в цепи, но и регулировать путем изменения силы тока.
3. Скорость электрохимического процесса зависит от ЭДС и существенно зависит от условий диффузии ионов. Диффузия ионов часто оказывает определяющее влияние на скорость электродного процесса.
Равновесное состояние электрода можно охарактеризовать следующими признаками:
1. Скорость протекания электрохимической реакции в анодную и катодную стороны одинакова на равновесном электроде как в целом, так и на любом его участке, достаточно большом по сравнению с размерами молекул. В состоянии равновесия
Ia = Ik = Io и ia = ik = io,
где Ia и Ik – токи, а ia и ik – плотности тока, отвечающие анодному и катодному процессам; Io и io – ток обмена и плотность тока обмена соответственно.
2. В состоянии равновесия не происходит никаких изменений ни в качественном, ни в количественном составе системы. Природа и масса электрода, а также состав электролита сохраняются неизменными.
3. В условиях установившегося равновесия заряд металла по отношению к раствору, а следовательно, и потенциал электрода не являются функцией времени; они определяются лишь составом системы, её температурой и давлением. Потенциал электрода в этих условиях называется обратимым или равновесным электродным потенциалом. Величину равновесного электродного потенциала можно вычислить при помощи общих термодинамических уравнений.
Неравновесная электрохимическая система, то есть система, в которой электрохимические превращения протекают в заданном направлении с конечной скоростью, отличается следующими признаками:
1. Скорость электрохимической реакции в анодном и катодном направлениях неодинакова: Ia ¹ Ik ¹ Io. В неравновесной электрохимической системе одна из двух возможных электродных реакций протекает преимущественно в анодном направлении, а другая – преимущественно в катодном направлении.
2. Масса (часто и природа) электрода, а также состав раствора вблизи него оказываются изменёнными по сравнению с состоянием равновесия.
3. Потенциал электрода под током Е i в общем случае не равен равновесному электродному потенциалу Ер и его нельзя вычислить термодинамически. Значение его зависит не только от природы системы, температуры и давления, но и от силы тока. Следовательно, и напряжение Е i для неравновесных электрохимических систем отличается от обратимой ЭДС Е. Напряжение гальванических элементов при этом меньше, а напряжение на электрохимической ванне больше, чем ЭДС.
Неравновесный электродный потенциал при достижении стационарности процесса может оказаться, подобно равновесному электродному потенциалу, практически не зависящим от времени. Это установившееся значение потенциала электрода под током называется стационарным потенциалом.
Таким образом, для неравновесной электрохимической системы должна существовать определённая связь между силой тока и скоростью протекающего в ней химического превращения, а также между силой тока и напряжением Е i (или потенциалами входящих в систему электродов).
Скорость электрохимической реакции v, подобно скорости химической реакции, определяется как количество вещества, изменившееся за единицу времени:
v = ± 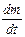 .
.
Поскольку между количеством прореагировавшего вещества и количеством прошедшего электричества существует прямая пропорциональность, то на основании первого закона Фарадея модно записать
v = ± = kэ 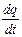 = kэ I,
= kэ I,
то есть скорость электрохимической реакции пропорциональна силе тока I. Так как для каждой данной реакции kэ постоянно, то сила тока представляет собой удобную величину для выражения скорости любого электрохимического превращения.
Характерной особенностью электрохимических реакций является то, что они совершаются на границе раздела электрод – электролит, и поэтому их скорость зависит от площади поверхности раздела S. В связи с этим принято относить скорость электрохимической реакции к единице поверхности раздела и определять ее как плотность тока:
i = I/S.
[ i ] = А/м2 (СИ); А/дм2 (в прикладной электрохимии); А/см2 (в ранних работах по теоретической электрохимии).
На основании законов Фарадея можно подсчитать, какое количество электричества потребуется для получения необходимого количества продукта электрохимической реакции; при 100%-ном выходе по току для получения 1 г-экв любого вещества требуется одно и то же количество электричества, равное одному Фарадею. Следует подчеркнуть, что законы Фарадея определяют расход количества электричества, но не электрической энергии; расход энергии зависит от природы электрохимической реакции и от условий ее протекания (от этого зависит напряжение на ванне Е).
Равновесие между раствором и электродом, имеющим определенный потенциал, является динамическим: происходит непрерывный обмен заряженными частицами между электродом и раствором. При равновесии скорости перехода частиц в противоположных направлениях одинаковы. Количество электричества, переходящее в этих условиях в единицу времени от электрода к раствору и обратно, называется током обмена. Существование тока обмена можно доказать методом изотопных индикаторов.
При прохождении электрического тока через границу электрод – раствор двухсторонний ток обмена имеется, но на него накладывается, как правило, несравненно больший односторонний ток, определяемый ЭДС элемента или приложенной внешней разностью потенциалов.
Электрический ток вызывает изменения на поверхности электродов, зависящие от многих факторов и прежде всего от силы тока. Изменение электрического состояния электрода (его потенциала, плотности заряда ДЭС) под влиянием проходящего через границу раздела электрического тока называется поляризацией электрода. При поляризации потенциал электрода изменяется по сравнению с тем «равновесным» значением, которое он имел в данном растворе при отсутствии тока:
DЕ = Е i – Еp,
DЕ – электродная поляризация; Е i – потенциал электрода «под током»; Еp – равновесный электродный потенциал. Так как при наложении катодного тока потенциал смещается в отрицательную сторону, а при наложении анодного – в положительную, то катодная электродная поляризация всегда отрицательна, а анодная всегда положительна:
DЕк = Е i – Еp < 0; DЕа = Е i – Еp > 0.
Любой электродный процесс представляет собой сложную гетерогенную реакцию, состоящую из ряда последовательных стадий. По крайней мере на некоторых из них она может протекать по двум или нескольким параллельным путям. Природа и число стадий каждой электрохимической реакции зависят от ее характера.
Из химической кинетики известно, что скорость последовательной реакции определяется скоростью наиболее медленной из ее последовательных стадий, а из ряда параллельных путей наиболее вероятен путь с наименьшими торможениями. Эти же представления справедливы и в случае электрохимических процессов. Стадия, определяющая скорость всего электродного процесса, называется замедленной или лимитирующей. Замедленность той или иной стадии является непосредственной причиной поляризации электрода. Если известна природа замедленной стадии, то есть ясна причина, обусловливающая появление поляризации, то вместо термина «поляризация» употребляют термин (электродное) перенапряжение h. Таким образом, перенапряжение – это поляризация электрода, обусловленная замедленным протеканием вполне определенной стадии суммарного электродного процесса.
В зависимости от природы замедленной стадии можно говорить о различных видах перенапряжения. Одной из обязательных стадий любого электродного процесса является транспортировка участников реакции – доставка (или отвод) к границе раздела электрод – электролит. Поляризацию, вызванную торможением на стадии транспортировки, называют концентрационной поляризацией, перенапряжением транспортировки или диффузионным перенапряжением hд . Замедленное протекание чисто химической стадии – реакции, предшествующей или следующей за актом разряда – вызывает появление химического или реакционного перенапряжения hх (hр). Любой электродный процесс включает в себя хотя бы одну стадию, связанную с переходом электронов через границу раздела электрод – электролит. Электродную поляризацию, вызванную замедленным протеканием этой стадии, называют электрохимическим перенапряжением hэ , поскольку именно стадия перехода электронов является собственно электрохимическим актом. Для описания этого вида перенапряжения широко используют также термины перенапряжение замедленного разряда, перенапряжение переноса заряда, перенапряжение (электронного) перехода. Наконец, замедленность стадии построения или разрушения кристаллической решетки, а также замедленность перехода от одной модификации к другой соответствуют фазовому перенапряжению hф .
В общем случае смещение потенциала электрода под током от равновесного значения представляет собой результат наложения всех видов перенапряжения:
DЕ = hд + hр + hэ + hф.
Однако можно найти такие электродные процессы и создать такие условия, при которых преобладающее значение будет иметь какой-либо один вид перенапряжения.
Концентрационная поляризация
Концентрационная поляризация обусловлена уменьшением в процессе электролиза концентрации ионов, определяющих потенциал у поверхности электрода; в результате этого изменяется равновесный потенциал электрода. Влиянием концентрационной поляризации на потенциал электрода под током можно пренебречь лишь при малых плотностях тока (то есть при малых скоростях электрохимической реакции). Напротив, при высоких плотностях тока стадии доставки могут определять скорость всего электродного процесса.
Рассмотрим процесс электролиза раствора AgNO3 с концентрацией с о (моль/л) в присутствии значительного количества KNO3 . Катод – серебряная проволока, анод – Pt-жесть с очень большой поверхностью. В отсутствие тока потенциал катода может быть вычислен по уравнению Нернста
Е = Ео + ln c о.
Приложим к электродам небольшую разность потенциалов. На катоде начнется восстановление ионов серебра в металлическое серебро. При прохождении тока концентрация ионов серебра в непосредственной близости у катода уменьшается, а концентрация их в остальной части раствора остается постоянной. Возникает некоторый градиент концентраций, вызывающий диффузию ионов из объема раствора к поверхности электрода, а электрод принимает потенциал Е i, соответствующий новому значению концентрации с S у его поверхности:
Е i = Ео + ln с S.
По мере прохождения тока градиент концентрации у катода увеличивается, и подача ионов из глубины раствора путем диффузии усиливается. Через некоторое время создаются такие условия, при которых количество разряжающихся ионов становится равным количеству ионов, которое подводится к поверхности электрода. Устанавливается некоторое стационарное (то есть не изменяющееся во времени) распределение ионов у катода.
В стационарных условиях сила тока, проходящего через раствор, определяется количеством грамм-ионов ni, продиффундировавших к электроду в единицу времени. Согласно закону Фика это количество равно
ni = DS 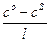 ,
,
D – коэффициент диффузии разряжающегося иона;
S – площадь поверхности электрода;
l – толщина диффузионного слоя (слоя, в котором происходит уменьшение концентрации от с о до с S).
Чтобы вычислить силу тока I, текущего к электроду, необходимо величину ni умножить на zF, где z – заряд разряжающегося иона, F – число Фарадея (для AgNO3 z = 1):
I = zF DS .
При увеличении силы тока величина с S уменьшается и при достижении некоторого предельного значения силы тока, называемого предельным током диффузии Iд , становится равной нулю. Поэтому предельный ток диффузии равен
Iд = zF DS 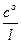 .
.
Из двух предыдущих уравнений также получим
с S = с о 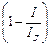 .
.
Подставив полученное выражение для с S в уравнение для Е i и вычтя из результата уравнение для Е, найдем, что сдвиг потенциала, обусловленный концентрационной поляризацией, будет равен
DЕ = 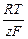 ln .
ln .
Величины DЕ обоих электродов складываются в ЭДС концентрационной поляризации, направленную против приложенной к электролитической ванне разности потенциалов, поэтому последняя должна быть увеличена на ЭДС концентрационной поляризации, чтобы была получена необходимая для электролиза сила тока. Так как в электрохимических производствах при электролизе применяют токи большой плотности, возникают значительные ЭДС поляризации, что увеличивает расход электрической энергии, поэтому устранение или уменьшение концентрационной поляризации является важной практической проблемой. Одной из основных мер уменьшения концентрационной поляризации является перемешивание растворов. Возникновение концентрационной поляризации снижает ЭДС химических источников тока при их работе. Избежать этого удается путем создания особых условий эксплуатации источников тока или применения насыщенных растворов солей с избытком твердой соли (элемент Вестона).
Электрохимическое перенапряжение
Любой электродный процесс обязательно включает в себя одну или несколько стадий, на которых частицы либо присоединяют к себе электроны (акт восстановления), либо теряют их (акт окисления). Однако сущность собственно электрохимической стадии не сводится только к изменению валентного состояния частицы или только к переносу заряда через границу раздела электрод–электролит. Приобретение (или потеря) частицей электрона приводит одновременно к изменению ее физико-химического и энергетического состояния. Так, например, в ходе реакции
Н3О+ + е – = Надс + Н2О
приобретение электрона частицей Н3О+ означает не только изменение заряда от величины z1 = 1 до величины z2 = 0, но и превращение гидратированного протона в адсорбированный на электроде атом водорода, то есть разрыв связей между ионом водорода и растворителем и создание связи между атомом водорода и металлом.
При осаждении металла из раствора его простой соли металлический ион из аквакомплекса переходит в состояние адатома (или адиона)
Mz+×aq + z e – = Мадс + Н2О
с последующим его вхождением в решетку металла.
Таким образом, приобретение или потеря частицей заряда всегда сопровождаются перестройкой ее структуры и изменением ее природы. Чем глубже эти изменения, тем больше должна быть энергия активации и тем ниже скорость собственно электрохимической стадии, то есть тем вероятнее, что именно она определяет скорость всего электродного процесса и обусловливает появление электрохимического перенапряжения.
Теория электрохимического перенапряжения была разработана применительно к процессу катодного выделения водорода, а затем распространена на другие электродные процессы. Первая попытка количественного оформления теории замедленного разряда была предпринята Эрдей-Грузом и Фольмером в 1930 г. Эрдей-Груз и Фольмер вывели формулу, связывающую потенциал электрода под током с плотностью тока. Выведенная ими формула является основным уравнением электрохимического перенапряжения и согласуется с эмпирическим уравнением для перенапряжения водорода. Однако теория замедленного разряда в ее первоначальном виде содержала ряд недостаточно обоснованных допущений и не могла удовлетворительно описать всю совокупность опытных данных. Наибольший вклад в теорию замедленного разряда был внесен А.Н.Фрумкиным (1933), который впервые учел влияние строения ДЭС на кинетику электрохимических процессов. Его идеи во многом определили основное направление развития электрохимической науки и ее современное состояние.
Напряжение разложения
Минимальная разность потенциалов, которую необходимо создать между электродами, чтобы электролиз начался, называется напряжением разложения электролита. При отсутствии перенапряжения на электродах напряжение разложения равно сумме равновесных потенциалов электродов, образующихся после начала электролиза (например, при электролизе НCl равно сумме равновесных потенциалов хлорного и водородного электродов). При наличии же перенапряжения хотя бы на одном электроде напряжение разложения больше суммы равновесных потенциалов.
Если при электролизе на электродах образуются твердые или жидкие растворы, и особенно при выделении газов, напряжение разложения зависит от формы и размеров электродов, характера их поверхности, условий удаления газов и многих других обстоятельств. Поэтому величина напряжения разложения не может служить однозначной характеристикой электролита при различных условиях.
Оказалось, что напряжения разложения при электролизе кислородсодержащих кислот и щелочей средних концентраций на платиновых электродах близки по своим значениям. По-видимому, при электролизе целого ряда веществ протекают одинаковые процессы как на катоде, так и на аноде. Действительно, в этих растворах на катоде выделяется водород, а на аноде – кислород. В растворах кислот разряжаются ионы гидроксония
2Н3О+ + 2 е = 2Н2О + Н2
В растворах щелочей также происходит разряд ионов гидроксония, а не ионов щелочного металла. Однако вследствие незначительной концентрации Н3О+ при большой силе тока водород в щелочи выделяется путем непосредственного разложения молекул воды, адсорбированных на электроде:
2Н2О + 2 е = 2ОН– + Н2
В растворах щелочей и солей щелочных металлов присутствуют два сорта катионов (например, К+ и Н3О+). При изменении потенциала электрода в отрицательную сторону вначале достигается потенциал разряда ионов гидроксония, который в нейтральном растворе при р = 1 атм равен всего – 0,4 В, и начинается процесс выделения водорода. Разряд же ионов калия (см. таблицу стандартных электродных потенциалов) может происходить лишь при высоких отрицательных потенциалах (при 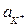 = 1 лишь при потенциале – 2,9 В), что при электролизе водных растворов вообще невозможно, так как при гораздо меньшем напряжении (1,3 – 1,7 В) начинается выделение Н2 и О2.
= 1 лишь при потенциале – 2,9 В), что при электролизе водных растворов вообще невозможно, так как при гораздо меньшем напряжении (1,3 – 1,7 В) начинается выделение Н2 и О2.
Выделение кислорода на аноде из щелочных растворов может быть следствием единственно возможной электродной реакции – разряда ионов гидроксила
4ОН– – 4 е = 2Н2О + О2
При электролизе кислот, где концентрация ионов гидроксила очень мала, кислород выделяется в результате непосредственного разложения молекул воды на аноде
6Н2О – 4 е = 4Н3О+ + О2
Ранее предполагалось, что в растворах кислородсодержащих кислот или их солей разряжаются соответствующие анионы. Это предположение неправильно. Ионы ОН– обладают наименьшим потенциалом разряда (+ 1,23 В), а потому при электролизе указанных солей выделение О2 обусловлено разрядом ионов ОН–.
Таким образом, при электролизе кислородсодержащих кислот, щелочей и соответствующих солей щелочных и щелочноземельных металлов на электродах протекает единственный процесс разложения воды, то есть выделение кислорода и водорода. Роль остальных ионов сводится лишь к обеспечению достаточной для электролиза электропроводности. Следует отметить, что близость напряжения разложения при электролизе кислот и щелочей наблюдается только при использовании электродов из определенных металлов (Pt, Pd), на которых мало перенапряжение водорода.
В растворах солей металлов менее электроотрицательных, чем водород, на катоде может выделяться уже металл. При электролизе кислот, не содержащих кислорода, и их солей на аноде, как правило, разряжаются соответствующие анионы.
Интересно поведение соляной кислоты. В концентрированных растворах на аноде выделяется хлор, а в разбавленных – кислород, причем меняется величина напряжения разложения. С разбавлением кислоты уменьшается активность ионов хлора, и равновесный потенциал хлорного электрода делается более положительным, чем потенциал разряда ионов ОН–, поэтому и происходит изменение анодного процесса: существенно уменьшается разряд ионов хлора и происходит разряд ионов гидроксила или молекул воды и выделение кислорода.
Лекция 60
Перенапряжение выделения водорода. Уравнение Тафеля. Теории водородного перенапряжения
Электролиз может начаться после того, как приложенная извне разность потенциалов достигает величины, равной (вернее, очень незначительно превышающей) разности обратимых потенциалов электродов электрохимической ячейки (потенциалы разряда ионов). Однако во многих случаях для того, чтобы электролиз начался, необходимо приложить к электролитической ванне разность потенциалов, на конечную величину большую, чем разность равновесных потенциалов электродов, образующихся при электролизе. Эта минимальная величина приложенной извне разности потенциалов называется напряжением разложения. То избыточное напряжение, которое необходимо приложить к ванне сверх ее равновесной ЭДС, чтобы начался электролиз, включает в себя, во-первых, перенапряжение на электродах (на катоде и на аноде), а во-вторых, омическое падение напряжения в растворе, соответствующее электросопротивлению этого раствора.
Величина перенапряжения на электроде зависит от природы электрода, плотности тока, состава раствора и других факторов. Величина перенапряжения различна для разных электрохимических процессов.
В связи с большим практическим значением реакции выделения водорода для ряда технических процессов (электролиз воды, хлорный электролиз, эксплуатация аккумуляторов и гальванических элементов, коррозия) эта электрохимическая реакция изучена наиболее детально.
|
|
|
|
|
Дата добавления: 2014-01-07; Просмотров: 2385; Нарушение авторских прав?; Мы поможем в написании вашей работы!