КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
А. Критерии моногенного наследования
|
|
|
|
1) Аутосомно-доминантный (АД) тип наследования (рис.3; примеры: с-м Марфана, хорея Гентингтона, миотония Томсена, прогрессирующая мышечная дистрофия (ПМД) лицелопаточноплечевая и др.):
- характерна передача патологического признака по вертикали, т.е. из поколения в поколение;
- вероятность передачи мутации от больного родителя потомству составляет 50%;
- оба пола поражаются с одинаковой частотой;
- больной отец может передать патологию сыновьям;
- если оба родителя здоровы (при исключении субклинических проявлений, обусловленных неполной пенетрантностью), но в семье рождается больной ребенок с АД синдромом, то такие случаи обусловлены новыми мутациями в родительской гамете, повторный риск заболевания в семье низкий;
- мутации в гомозиготном состоянии (встречаются крайне редко), как правило летальны.
 рис.3
рис.3
 |




 I 1 2 3 4 5
I 1 2 3 4 5
 | |||||||||||||
 |  |  | |||||||||||
 |  | ||||||||||||
 | |||||||||||||
II









 1 2 3 4 5 6 7 8 9 10
1 2 3 4 5 6 7 8 9 10




 |
III














 1 2 3 4 5 6 7 8 9 10
1 2 3 4 5 6 7 8 9 10
 |
2) Аутосомно-рецессивный (АР) тип наследования (рис.4; примеры: атаксия Фридрейха, спинальная мышечная атрофия 5q, фенилкетонурия, адреногенитальный с-м и др.):
- действие рецессивного признака проявляется в гомозиготном состоянии;
- у здоровых родителей с вероятностью 25% рождаются больные дети, что подтверждает гетерозиготное носительство родителей;
- с одинаковой частотой поражаются оба пола;
- если болен один из родителей, то, как правило, все потомки рождаются здоровыми, но в 100% гетерозиготными носителями;
- в браке между больным и гетерозиготным носителем повторный риск достигает 50% - так называемое "псевдодоминантное наследование", наблюдающееся очень редко.
- среди семей с АР формами моногенных болезней (МБ) чаще встречаются кровнородственные браки.
Предполагается, что каждый человек в любой популяции является носителем от 5 до 10 рецессивных мутаций, находящихся в гетерозиготном состоянии.
рис.4 



 1 2 3 4
1 2 3 4





 I
I
 |  |





 1 2 3 4 5 6 7 8
1 2 3 4 5 6 7 8




 II
II
 |





 1 2 3 4 5 6 7 8 9 10
1 2 3 4 5 6 7 8 9 10
 III
III




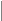



 IV 1 2 3 4 5 6
IV 1 2 3 4 5 6
3) Х-сцепленный рецессивный (ХР) тип наследования (рис.5; примеры: ПМД Дюшенна/Беккера, гемофилия, болезнь Кеннеди и др.):
- чаще всего, патологический признак передается через здоровых женщин, являющихся гетерозиготными носительницами, половине своих сыновей (т.е. суммарный повторный риск в такой ситуации составит 25%);
- все дочери от матерей-носительниц фенотипически здоровы, но половина из них будут являться такими же гетерозиготными носительницами;
- заболевание может прослеживаться у мужчин - родственников пробанда по материнской линии;
- в случае, если болен отец, то все его потомство рождается здоровым, однако 100% дочерей будут являться гетерозиготными носительницами;
- если женщина не является носительницей, но в семье рождается больной мальчик, то это можно объяснить новой мутацией, происшедшей в материнской половой клетке;
- в редких случаях (при сочетании с синдромом 45,ХО, а также в браке между пораженным мужчиной и женщиной - гетерозиготной носительницей) заболевание может наблюдаться у девочек.
 рис.5
рис.5
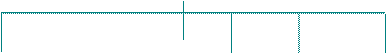 |
I

 1 2 3 4 5
1 2 3 4 5
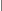 | |||||||||||||||
| |||||||||||||||
| | | |||||||||||||
| |  | |||||||||||||
II
 1 2 3 4 5 6 7 8 9 10
1 2 3 4 5 6 7 8 9 10
 |  | | | |||||||||||||
| | | |  | | |||||||||||
 III
III 1 2 3 4 5 6 7 8 9 10
1 2 3 4 5 6 7 8 9 10
 |  |  | | ||||||
 |
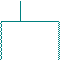
4) Х-сцепленный доминантный (ХД)тип наследования (рис.6; примеры: вариан- ты болезни Штрюмпеля, невральной амиотрофии Шарко-Мари-Тус, витамин-Д-резистентный рахит и др.):
- поражаются как мужчины так и женщины, однако женщины чаще, примерно в два раза;
- больная мать передает признак половине своих сыновей и дочерей, т.е. повторный риск для потомства составляет 50%;
- больной отец также с вероятностью 50% передает патологию, однако все его сыновья будут здоровы, а все дочери - больны;
- впервые возникший случай заболевания в семье обусловлен новой мутацией в родительских гаметах.
рис.6
 | |||
|
 I 1 2 3 4 5
I 1 2 3 4 5
| |||||||||||
| |  | |||||||||
| | ||||||||||

 1 2 3 4 5 6 7 8 9 10
1 2 3 4 5 6 7 8 9 10
 II
II
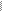

 1 2 3 4 5 6 7 8 9 10
1 2 3 4 5 6 7 8 9 10
III
|
5) Y сцепленное наследование (рис.7)
- если признак передается от отца, то он проявляется только у сыновей в 100% случаев.
рис.7
 1 2
1 2

 I
I
 |



 1 2 3 4 5
1 2 3 4 5



 II
II
 |  |  | |||||||
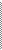 | 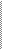 |



 1 2 3 4 5 6 7 8 9
1 2 3 4 5 6 7 8 9
III
6) Материнское наследование, обусловлено мутациями в митохондриальной ДНК (рис.8; примеры: митохондриальные миопатии, синдром Кернс-Сейра и др.)
- при передаче патологического признака от матери болеет с разной степенью тяжести все потомство;
- если болен отец, то все дети рождаются здоровыми;
- впервые возникший случай заболевания в семье обусловлен новой мутацией.
рис.8
 1 2
1 2

 I
I
 |

 1 2 3 4 5
1 2 3 4 5

 II
II
| |||||||||
 |  |  | |
 III 1 2 3 4 5 6 7 8 9 10
III 1 2 3 4 5 6 7 8 9 10
Б) Критерии полигенного наследования. Полигенное наследование имеет место при мультифакториальных болезнях (МФБ); примеры: язвенная болезнь, сахарный диабет, атеросклероз, ишемическая болезнь сердца, гипертоническая болезнь, шизофрения и др.), т.е. при заболеваниях в возникновении которых играют роль как наследственные так и средовые факторы. При МФБ патологические признаки не имеют менделевского характера передачи. Повторный риск заболевания в семьях рассчитывается эмпирически с учетом следующих факторов:
- степени родства с пораженным:
I ст. - родители - дети, сибсы - 50% общих генов;
II ст. - тети, дяди - племянники; дедушки, бабушки - внуки; двоюродные сибсы - 25% общих генов;
III ст. - троюродные сибсы; прадедушки, прабабушки - правнуки; 12,5% общих генов;
IV ст. - четвероюродные сибсы - 6,25% общих генов;
- повторный риск зависит от количества пораженных в семье (т.е. от семейного накопления случаев болезни): 1) если родители здоровы, а у пробанда имеются признаки болезни, то риск для его сибсов составит от 5 до 10%; 2) если болен один из родителей и пробанд, то риск для сибсов составит от 10 до 20%, а для детей пробанда ~ 10%; 3) если больны оба родителя и пробанд, риск для сибсов составит от 20 до 40%, 20% - для детей пробанда;
- повторный риск зависит от формы заболевания, возраста начала, тяжести течения;
- имеет значение количество вовлеченных в патологический процесс генов, т.е. фактор наследуемости;
- принимается во внимание близнецовый критерий;
- повторный риск возрастает при передаче признака через редко поражаемый пол;
- учитывается сегрегационное отношение пораженных и непораженных сибсов в браках.
Таким образом расчет повторного риска при МФБ задача непростая и требует детального анализа анамнестических и генеалогических данных. В медико-генетических консультациях существуют таблицы в которых указывается эмпирические значения риска при конкретных заболеваниях.
5) Следующий этап клинико-генеалогического обследования - осмотр пробанда и его родственников (недообследование родственников может внести ошибочное мнение о типе наследования заболевания, т.к. нередко родственники считают себя здоровыми при наличии мягких или субклинических проявлений болезни). Клиническое обследование пораженных в семье основывается на синдромологическом подходе, суть которого заключается в подробнейшем сборе микро- и макропризнаков заболевания с последующим установлением конкретного синдрома. Пример: синдром Барде-Бидля складывается из следующих симптомов: ожирение, умственная отсталость, полидактилия, катаракта.
II. Семиотика наследственных болезней.
Рассмотрим семиотику наследственных и врожденных заболеваний. СЕМИОТИКА - это учение о синдромах и симптомах болезни. При проведении адекватного медико-генетического консультирования, эффективность которого в целом зависит от точно установленного диагноза, очень важно знать признаки наследственных болезней. Рассмотрим общие свойства этой патологии:
1) Ранний период манифестации: 25% моногенных болезней проявляются с рождения; к 3-х летнему возрасту - 70%; до завершения пубертатного возраста - 90%. Однако известны формы НБ, при которых отмечается поздняя манифестация, например: поликистоз почек (АД-вариант), хорея Гентингтона проявляются, чаще всего, после 30 лет; болезнь Альцгеймера - после 50-60 лет;
2) Разнообразие вариантов первичных генетических дефектов и их типов наследования: существуют моногенные болезни, обусловленные генными мутациями и передающиеся по АД, АР, Х-сцепленному, Y-сцепленному, материнскому наследованию; хромосомные болезни (не имеющие классического менделевского наследования), при которых мутации захватывают группы сцепления генов; мультифакториальные болезни с полигенным наследованием.
3) Множественность (полиорганность) поражения, обусловленное, в основном, плейотропным эффектом гена (т.е. один ген обуславливает формирование нескольких признаков).
Примеры: при синдроме "рука-сердце" (синдром Холт-Орама, АД) имеет место выраженный клинический полиморфизм, при котором экспрессивность признаков варьирует от атрезии (отсутствия) лучевой кости и порока сердца в виде дефекта межпредсердной, межжелудочковой перегородок до гипоплазии 1-го пальца и пролапса митрального клапана. Кроме того, могут иметь место различные скелетные аномалии: отсутствие большой грудной мышцы, расщепление неба. Интеллект при этой форме нормальный. Ген локализован предположительно в сегменте 14q23-q24.2.
- Синдром ожирения и умственной отсталости (синдром Лоуренса-Муна, с-м Барде-Бидля): характеризуются умственной отсталостью, гипогенитализмом, пигментной ретинопатией, возможно наличие нижней спастической параплегии, полидактилии, часто имеет место судорожный синдром, аномалии развития мозга. Тип наследования - Аутосомно-рецессивный (АР).
4) Накопление больных либо характерных проявлений в семье (сегрегация). Пример: в одном из районов Мордовии выявлена семья, в которой диагноз НАШМТ установлен у 30 родственников.
5) Хронический и прогредиентный характер течения. Типичен "светлый" промежуток до начала заболевания (особенно для наследственных болезней обмена веществ).
Пример: лейкодистрофии. Различают несколько форм: глобоидноклеточная (болезнь Краббе, АР), метахроматическая лейкодистрофия Пилециуса-Мерцбахера (ХР) и др.. Рассмотрим метахроматическую лейкодистрофию, которая, в свою очередь, подразделяется на три варианта: 1) позднюю инфантильную (продолжительность жизни - до 5 лет); 2) ювениальную (начало - от 4 до 10 лет с медленно прогрессирующим течением); 3) форму взрослых с началом после 16 лет, характеризующуюся психическими изменениями, постепенно нарастающей деменцией, на основании чего больным иногда выставляется диагноз "шизофрения". Основной биохимический дефект обусловлен нарушением деградации сульфатидов с накоплением их в ЦНС, сетчатке и внутренних органах. Патоморфологически определяется диффузная демиелинизация структур ЦНС. Частота - 1:40000, тип наследования - АР, ген локализован на 22q13.31-qter.
6) Резистентность к терапии. Пример: Наследственные мото-сенсорные невропатии (НАШМТ) устойчивы к лечебному воздействию, однако это не абсолютный показатель (например, иногда имеет место стойкая ремиссия после санаторно-курортного лечения, т.е. требуется индивидуальный подход к подбору методов терапии, постоянный поиск эффективных способов лечения). Поэтому не следует отказывать больным в лечении, особенно если пациент настроен на терапию. При ряде форм (болезнь Вильсона-Коновалова, фенилкетонурия, врожденный гипотиреоз и др.) своевременное проведение терапии приносит ощутимые положительные результаты. Кроме того, классические методы лечения и реабилитации больных с наследственной патологией нередко существенно затормаживают прогрессирование, инвалидизацию, что положительно сказывается на психологической и социальной адаптации пациентов и их семей. В настоящее время разрабатывается более 400 протоколов генотерапии НБ, соответственно, эффективность генотерапевтического воздействия будет зависеть, во многом, от степени инвалидизации пациента.
7) Наличие микро- и макроаномалий развития. Микроаномалия (МА) развития - это дефект органа или системы, который не изменяет функцию и не является выраженным косметическим дефектом. Макроаномалия (МКА), соответственно, нарушает функцию органа или системы и может выражаться в виде грубого косметического дефекта.
Микроаномалии довольно часто встречаются в популяции и необязательно при наследственной патологии: сандалевидная щель между 1 и 2 пальцами стоп, частичная синдактилия (сращение) между 2 и 3 пальцами стоп, клинодактилия - признаки часто встречающиеся. Поперечная ладонная складка может иметь место без сочетания с НБ. Как у пораженных, так и у здоровых людей МА могут изменяться и даже исчезать с возрастом, т.к. формирование организма происходит до окончания пубертатного периода. У людей с НБ микроаномалии встречаются гораздо чаще. Если у пациента выявляется 5 и более МА (условный критерий), то следует задуматься на предмет наследственной патологии.
8) Нарушение физического, психического, полового развития и функции репродукции; снижение продолжительности жизни и повышенная летальность.
9) Выраженный клинический полиморфизм, т.е. разнообразие проявлений, что обусловлено, с одной стороны, генетической гетерогенностью НБ (т.е. клинически сходные признаки детерминированы различными мутациями), с другой - воздействием внешнесредовых факторов.
В клинической генетике используется синдромологический подход к установлению диагноза. На первом этапе тщательно выявляются симптомы болезни, формирующие определенный синдром, что позволяет, после дополнительного лабораторно-инструментального обследования пациента, перейти к формулировке окончательного диагноза.
- Пример: при с-ме Корнелии де Ланге имеет место задержка психомоторного развития, микроцефалия, синофриз, длинный фильтр, "вывернутые" кнаружи ноздри, тонкая "завернутая" внутрь верхняя губа, гипертрихоз, отставание в росте, микромелия (гипоплазия предплечий).
- Пример: при лопаточно-перонеальной амиотрофии Старка-Кайзера (АД) выявляются атрофии перонеальных, лопаточных мышц, диффузные фасцикулляции и фибрилляции в мышцах, тремор пальцев рук, деформации стоп, изменение походки по типу "степпажа".
Однако, не все НБ имеют типичные проявления. Так диагностика ряда хромосомных болезней невозможна только на основе фенотипа и требует цитогенетического исследования
Рассмотрим микро- и макроаномалии, которые могут наблюдаться при НБ со стороны различных систем организма.
1. Изменение роста.
1) Низкий рост. Чаще имеет место задержка роста, которая может быть: а) пропорциональной - родители, как правило, обращаются к эндокринологам; пример: пангипопитуитарная карликовость;
б) диспропорциональной, которая подразделяется на:
- короткоконечностную карликовость (за счет укорочения длинных трубчатых костей); брахиморфия - макросомия в сочетании с укорочением нижних конечностей;
- короткотуловищную (за счет нарушения роста туловища).
Примеры: с-м Нунан (АД), ахондроплазия (АД, укорочение, преимущественно, за счет проксимальных отделов конечностей) и др. Низкий рост характерен для хромосомных заболеваний.
2) Высокий рост. Примеры: с-м Марфана (АД), гомоцистинурия (АР), с-м Клайнфельтера (47, XXY), с-м 47,XYY и др.
2. Нарушения веса. Примеры: с-м Беквита-Видемана (пример макросомии), включающий следующие признаки - макросомию (пропорциональный избыток весоростовых показателей), вес при рождении более 5 кг., омфалоцеле (грыжа пупочного канатика), макроглоссия, дизэмбриомегалия (т.е. увеличение размеров внутренних органов); с-м Прадера-Вилли, характеризующийся появлением булимии на 2-м году жизни, быстрым развитием ожирения. Детская форма прогерии является примером низкого веса и роста (микросомии). При данной форме происходит атрофия подкожной жировой клетчатки, развитие атеросклеротических изменений в детском возрасте, преждевременное старение.
3. Изменения со стороны кожных покровов.
- Гиперпигментации и депигментации, генерализованные и локальные. Примеры: витилиго (АД), при котором выявляются ассиметричные, неправильной формы пятна депигментации; нейрофиброматоз (болезнь Реклинкгаузена), при котором выявляются множественные (>5) пятна цвета "кофе с молоком", веснусчатость в подмышечных и паховых областях, а также наличие множественных мягкоэластичных внутри-, подкожных фибром.
- влажность кожных покровов: ангидроз (отсутствие потоотделения), гипергидроз (избыточное потоотделение). Пример: ихтиоз (повышена сухость), ладонно-подошвенная кератодермия (повышено ороговение поверхностных слоев кожных покровов, особенно в области наружных поверхностей коленных, локтевых суставов, подошв, ладоней).
При некоторых формах изменяется эластичность кожи, в следствие патологии соединительнотканных элементов. Пример: с-м Элерса-Данлоса, при котором значительно возрастает растяжимость кожных покровов.
4. Изменения со стороны ногтей: гипо-, аплазия (полное отсутствие), ониходистрофия, анонихия (полное отсутствие ногтевых пластинок); пахионихия - утолщение ногтевых пластинок.
5. Изменения со стороны волос: гипо-, гипертрихоз, аллопеция (локальная и генерализованная), повышенная сухость, ломкость, упругость, гнездная седина и др.
6. Изменения со стороны черепа:
- микро-, макрокрания; микро-, макроцефалия (если окружность головы на 5 см. меньше возрастной нормы, то говорят о микроцефалии, наоборот - макроцефалии);
- брахицефалия - преобладание поперечных размеров окружности головы, выступающие лобные бугры, плоский затылок;
- гидроцефалия - имеет место несоответствие мозгового и лицевого отделов черепа с преобладанием мозгового; расширены подкожные вены, выбухают роднички, имеет место расширение желудочковой системы, субарахноидального пространства, выявляемое при проведении компьютерной томографии головного мозга;
- долихоцефалия - увеличение вертикальных размеров черепа;
- скафоцефалия - ладьевидная форма черепа с выступающими лобными и затылочными буграми;
- тригоноцефалия - треугольная форма черепа, с расширением в затылочной области;
- акроцефалия - заостренная по сагиттальному шву форма черепа;
- оксицефалия - башенный череп с заострением кверху ("сахарная голова");
- сфеноцефалия - расширение черепа в лобной и сужение в затылочной обл.;
- плагиоцефалия - ассиметрия мозгового черепа, связанная с неравномерным развитием разных частей черепа;
- грубые черты лица ("гаргоилизм") характерны для болезней накопления.
7. Изменения со стороны ушных раковин:
- макро-, микротия - увеличение и уменьшение в размере ушных раковин;
- насечки на мочке уха;
- преаурикулярные выросты, кисты;
- оттопыренные ушные раковины - часто встречающийся признак;
- гипо-, гиперплазия отдельных структур;
- околоушные фистулы;
- атрезия, стеноз наружных слуховых проходов (с-м Франческетти, АД).
В норме нижний край уха находится на линии, соединяющей основание сосцевидного отростка и крыла носа.
8. Область глаз:
- В норме между внутренними углами глаз помещается одна вертикально расположенная глазная щель;
- гипертелоризм - широкий промежуток между внутренними углами глаз (> одной вертикально расположенной глазной щели);
- гипотелоризм - малый промежуток между внутренними углами глаз (< одной вертикально расположенной глазной щели);
- монголоидный разрез глазных щелей - наружные углы глаз приподняты (синдром Дауна), глазные щели узкие;
- антимонголоидный - наружные углы глаз опущены (с-м Франческетти)
- необходимо учитывать этнические особенности;
- микро-, макрофтальм - соответственно уменьшенные и увеличенные в размере глазные яблоки, буфтальм или «бычий глаз» - крайнее проявление макрофтальма;
- анофтальм - отсутствие глазного яблока;
- энофтальм - глубоко посаженные глазные яблоки;
-экзофтальм - «пучеглазие», выступающие кнаружи увеличенные глазные яблоки (с-м Крузона, АД);
- микро-, макрокорнеа - соответственно уменьшенная и увеличенная в размере роговица;
- голубые склеры;
- колобома радужной оболочки, зрачка, века - арковидный дефект;
- гетерохромии радужной оболочки - различают полную гетерохромию (когда радужная оболочка одного глаза отличается по цвету от противоположной) и секторальную (внутри радужной оболочки имеется сектор другого цвета),
- аниридия - отсутствие радужной оболочки;
- изменения на глазном дне: атрофия диска зрительного нерва, симптом "вишневой косточки" (при наследственных болезнях обмена веществ);
- блефарофимоз - сужение глазных щелей;
- эпикант - вертикальная полулунная складка у внутреннего угла глаз (третье, добавочное веко);
- телекант - смещение внутренних углов глазных щелей латерально при нормальном расположении орбит;
- криптофтальм - сращение век;
- аномалии роста ресниц: дистихиаз - двойной ряд ресниц; тристихиаз - тройной ряд;
- афакия - отсутствие хрусталика;
- аблефария - отсутствие 1 или 2 век;
- анкилоблефарон - сращение краев век спайками;
- дискория - неправильная форма зрачка;
- коректопия - врожденное смешение зрачка;
- поликория - множественные зрачки;
- страбизм - косоглазие;
- эпитарзус врожденные складки коньюнктивы, идущие параллельно краю века и переходящие на другое веко;
9. Область носа:
- переносица: широкая, запавшая, сглаженная;
- гипоплазия крыльев носа, колобома крыльев носа (арковидный дефект), раздвоение кончика носа, укорочение или удлинение фильтра (фильтр - расстояние от кончика носа до края верхней губы).
10. Челюстная область:
- агнатия - отсутствие челюсти;
- прогнатия (выступающая верхняя челюсть), ретрогнатия (смещение верхней челюсти кзади);
- прогения, ретрогения (тоже по отношению к нижней челюсти);
- хейлоскиз - расщелина верхней губы - «заячья губа», чаще встречается при мультифакториальных заболеваниях.
11. Область рта и полости:
- макро-, микростомия - соответственно увеличенная и уменьшенная наружное отверстие ротовой полости;
- адентия - отстутствие зубов;
- олигодентия - количество зубов меньше нормального числа;
- диастема - широкая щель между центральными резцами;
- амелогенез (изменение цвета эмали);
- высокое, готическое, арковидное небо;
- палатоскиз - «волчья пасть» или расщелина неба, может быть мультифакториальным и моногенным признаком;
- "губы тапира" - большие, вывернутые кнаружи - наблюдается при ПМД Ландузи-Дежерина;
- макроглоссия - большой язык, иногда непомещающийся в ротовой полости (с-м Беквита-Видемана, ПМД Дюшенна/Беккера);
- анкилоглоссон - сращение языка.
12. Область шеи:
- короткая, широкая, кривошея; птеригиум - наличие кожных складок на боковых поверхностях (с-м Шерешевского-Тернера); низкий рост волос на задней поверхности; срединные, боковые кисты;
13. Область грудной клетки, позвоночника:
- различные деформации: уплощение, воронкообразное втяжение, атрезия мечевидного отростка, килевидная грудная клетка (при мукополисахаридозах) добавочные ребра, крыловидные лопатки, кифоз, кифосколиоз, гибус - горб (наблюдаются при различных формах миопатий, спинальных амиотрофий); гиперлордоз поясничного отдела позвоночника (при миопатиях); симптом "рыбьих позвонков" (двояковогнутая форма - при несовершенном остеогенезе);
- отсутствие большой грудной мышцы (с-м Холт-Орама);
- ателия (отсутствие сосков), полителия (наличие более 2-х сосков), гинекомастия - увеличение молочных желез у мужчин;
14. Область живота:
- грыжи пахово-мошоночные, пупочные, белой линии живота, стрии (при наследственной обменной патологии).
15. Аномалии строения половых органов:
- у мужчин: эписпадия (смещение наружного отверстия мочеиспускательного канала вверх), гипоспадия (смещение вниз); крипторхизм (нахождение яичка в кА….
одного или обоих яичек в следствие задержки опускания в мошонку); моноорхизм - наличие единственного яичка; макроорхизм - увеличение в размере яичек (при синдроме ломкой Х-хромосомы, с-ме 47,ХYY); анорхидия - 2-стороннее недоразвитие яичек;
- у женщин: атрезии, гипоплазии половых органов (с-м Шерешевского-Тернера, с-мы дисгенезии гонад, с-м тестикулярной феминизации и др.).
16. Аномалии конечностей:
- фокомелия - полное или частичное отсутствие проксимальных отделов конечностей;
- брахимелия - укорочение конечности;
- сиреномелия - сращение конечностей;
- долихостеномелия - резкое удлинение конечностей;
- аподин - отсутствие нижней конечности;
- апус - полное отсутствие обеих нижних конечностей;
- ахирия - отстутствие кисти;
- гемибрахия - отстутствие предплечья;
- зктромелия - отсутствие дистальной части конечности;
-брахидактилия - укорочение пальцев; изодактилия - расположение конечных фаланг на одном уровне; клинодактилия - изогнутые, скошеннные на конус пальцы; арахнодактилия ("пальцы паука") - длинные тонкие пальцы, утолщенные в обл. концевых фаланг; симфалангия - сращение межфаланговые; синдактилия (полная, частичная, кожная, костная); полидактилия, олигодактилия
- соответственно наличие добавочных или недостаток пальцев; эктродактилия - расщелины кистей, стоп в области пястных, плюстневых костей с атрезией структур 2-х, 3-х, 4-х пальцев, части пястных и плюстневых костей - «клешнеобразные конечности»;
- камптодактилия - сгибательные контрактуры проксимальных межфаланговых суставов кистей;
- сандалевидная щель (широкий промежуток между первым и вторым пальцами стоп); плоскостопие, полая стопа, стопа Фридрейха (при НАШМТ, атаксии Фридрейха); стопа-качалка (плоская с выступающей пяткой);
- ризомелическое укорочение конечностей - укорочение за счет проксимальных отделов - бедра, плеча (при ахондроплазии);
- мезомелическое укорочение конечностей - укорочение за счет голени, предплечья (при с-ме каудальной регресии).
- ДОПОЛНИТЬ!
В заключение рассмотрения раздела семиотики наследственных болезней необходимо подчеркнуть важность тщательного клинико-генеалогического обследования как больного, так и его родственников. Правильно проведенный генеалогический и синдромологический анализ является основой дальнейшей тактики обследования и, соответственно, лечения и профилактики при врожденных и наследственных заболеваниях, т.е. является залогом адекватого оказания квалифицированной медико-генетической помощи отягощенным семьям.
|
|
|
|
Дата добавления: 2014-01-11; Просмотров: 1769; Нарушение авторских прав?; Мы поможем в написании вашей работы!