КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Нарушение транспорта анионок хлоридорея 3 страница
|
|
|
|
Гиперурикемия, обусловленная частичной или полной недостаточностью гипоксантингуанинфосфорибозилтрансферазы, успешно поддается воздействию аллопуринола—ингибитора ксантиноксидазы. При этом у небольшого числа больных образуются ксантиновые камни, но большинство из них с почечными камнями и подагрой излечиваются. Специфических средств лечения при неврологических нарушениях при синдроме Леша—Найхана не существует.
Варианты ФРПФ-синтетазы. Выявлено несколько семей, у членов которых была повышена активность фермента ФРПФ-синтетазы (см. реакцию 3 на рис. 309-4). Все три известных типа мутантного фермента обладают повышенной активностью, что приводит к увеличению внутриклеточной концентрации ФРПФ, ускорению биосинтеза пуринов и усилению экскреции мочевой кислоты. Эта болезнь также наследуется как признак, сцепленный с Х-хромосомой. Как и при частичной недостаточности гипоксантингуанинфосфорибозилтрансферазы, при этой патологии на втором—третьем 10-летии жизни обычно развивается подагра и часто образуются мочекислые камни. У нескольких детей повышенная активность ФРПФ-синтетазы сочеталась с нервной глухотой.
Другие нарушения пуринового обмена. Недостаточность аденинфосфорибозилтрансферазы. Аденинфосфорибозилтрансфераза катализирует превращение аденина в АМФ (см. реакцию 4 на рис. 309-4). Первый человек, у которого была обнаружена недостаточность этого фермента, был гетерозиготным по этому дефекту, клиническая симптоматика у него отсутствовала. Затем было выяснено, что гетерозиготность по этому признаку распространена достаточно широко, вероятно, с частотой 1:100. В настоящее время выявлены 11 гомозигот по недостаточности этого фермента, у которых почечные камни состояли из 2,8-диоксиаденина. Из-за химического сходства 2,8-диоксиаденин легко спутать с мочевой кислотой, поэтому этим больным вначале был ошибочно поставлен диагноз мочекислого нефролитиаза.
Таблица 309-2. Структурные и функциональные нарушения при мутантных формах гипоксантингуанинфосфорибозилтрансферазы человека
| Мутантный фермент | Клинические проявления | Мутация | Функциональные нарушения | ||||
| замена аминокислоты | положение | внутриклеточная концентрация | максимальная скорость | константа Михаэлиса | |||
| гипоксантин | ФРПФ | ||||||
| ГФРТТоронто | Подагра | Arg-®Gly | Уменьшена | В пределах нормы | В пределах нормы | В пределах нормы | |
| ГФРТЛондон | » | Ser®Leu | » | То же | Повышена в 5 раз | То же | |
| ГФРТЭнн-Арбор | Нефролитиаз | Неизвестна | Н | » | »» | В пределах нормы | »» |
| ГФРТМюних | Подагра | Ser®Arg | В пределах нормы | Снижена в 20 раз | Повышена в 100 раз | »» | |
| ГФРТКинстон | Синдром Леша—Найхана | Asp®Asn | То же | В пределах нормы | Повышена в 200 раз | Повышена в 200 раз |
Примечание. ФРПФ означает 5-фосфорибозил-1-пирофосфат, Arg — аргинин, Gly — глицин, Ser — серин. Leu — лейцин, Asn — аспарагин. Asp— аспарагиновая кислота, ® — заменена (по Wilson et al.).
Недостаточность дезаминазы аденозина и недостаточность пуриннуклеозидфосфорилазы см. в гл. 256.
Недостаточность ксантиноксидазы. Ксантиноксидаза катализирует окисление гипоксантина в ксантин, ксантина в мочевую кислоту и аденина в 2,8-диоксиаденин (см. реакцию 8 на рис. 309-4). Ксантинурия, первое врожденное нарушение пуринового обмена, расшифрованное на ферментном уровне, обусловлена недостаточностью ксантиноксидазы. В результате у больных с ксантинурией выявляют гипоурикемию и гипоурикацидурию, равно как и усиленную экскрецию с мочой оксипуринов—гипоксантина и ксантина. Половина больных не предъявляют жалоб, а у 1/3 в мочевых путях образуются ксантиновые камни. У нескольких больных развилась миопатия, у трех — полиартрит, который мог быть проявлением вызванного кристаллами синовита. В развитии каждого из симптомов большое значение придают выпадению ксантина в осадок.
У четырех больных врожденная недостаточность ксантиноксидазы сочеталась с врожденной недостаточностью сульфатоксидазы. В клинической картине у новорожденных доминировала выраженная неврологическая патология, что характерно для изолированной недостаточности сульфатоксидазы. Несмотря на то что в качестве основного дефекта постулирована недостаточность молибдатного кофактора, необходимого для функционирования того и другого фермента, лечение молибдатом аммония было малоэффективным. У больного, находившегося полностью на парентеральном питании, развилось заболевание, симулирующее сочетанную недостаточность ксантиноксидазы и сульфатоксидазы. После проведенного лечения молибдатом аммония полностью нормализовалась функция ферментов, что привело к клиническому выздоровлению.
Недостаточность миоаденилатдезаминазы. Миоаденилат-дезаминаза, изофермент аденилатдезаминазы, обнаруживают только в скелетных мышцах. Фермент катализирует превращение аденилата (АМФ) в инозиновую кислоту (ИМФ). Эта реакция представляет собой составную часть пуринонуклеотидного цикла и, по-видимому, важна для поддержания процессов продукции и утилизации энергии в скелетной мышце.
Недостаточность этого фермента определяется только в скелетной мышце. У большинства больных при физической нагрузке появляются миалгии, мышечные спазмы и чувство утомления. Примерно 1/3 больных жалуются на мышечную слабость даже при отсутствии нагрузки. Некоторые больные жалоб не предъявляют.
Заболевание обычно проявляется в детском и подростковом возрасте. Клинические симптомы при нем те же, что и при метаболической миопатии. Уровень креатининкиназы повышен менее чем в половине случаев. Электромиографические исследования и обычная гистология мышечных биоптатов позволяют выявить неспецифические изменения. Предположительно недостаточность аденилатдезаминазы можно диагностировать на основании результатов теста на работоспособность ишемизированного предплечья. У больных с недостаточностью этого фермента продукция аммиака снижена, поскольку заблокировано дезаминирование АМФ. Диагноз следует подтверждать путем прямого определения АМФ-дезаминазной активности в биоптате скелетной мышцы, так как. сниженная продукция аммиака при работе характерна и для других миопатий. Заболевание прогрессирует медленно и в большинстве случаев приводит к некоторому снижению работоспособности. Эффективной специфической терапии не существует.
Недостаточность аденилсукциназы. Больные с недостаточностью аденилсукциназы отстают в психическом развитии и часто страдают аутизмом. Кроме того, они страдают судорожными припадками, у них задержано психомоторное развитие, отмечается и ряд двигательных расстройств. Экскреция с мочой сукциниламиноимидазолкарбоксамидрибозида и сукциниладенозина усилена. Диагноз устанавливают при обнаружении частичного или полного отсутствия активности фермента в печени, почках или скелетных мышцах. В лимфоцитах и фибробластах определяется его частичная недостаточность. Прогноз неизвестен, и специфического лечения не разработано.
ГЛАВА 310. ГЕМОХРОМАТОЗ
Лоури У. Пауэлл, Курт Дж. Иссельбахер (Lawrie W. Powell, Kurt J. Isselbacher)
Определение. Гемохроматоз представляет собой болезнь накопления железа, при которой чрезмерно увеличенное его всасывание в кишечнике приводит к его скоплению в тканях с последующим их повреждением и функциональной недостаточностью органов, особенно печени, поджелудочной железы, сердца и гипофиза. В 1889 г. фон Реклингхаузен назвал эту болезнь гемохроматозом, а железосодержащий пигмент — гемосидерином, поскольку он считал, что пигмент образуется в крови. Для того чтобы указать на присутствие окрашиваемого железа в тканях, часто используют термин «гемосидероз» или «сидероз», но для точной оценки состояния обмена железа в организме требуются его количественные определения в тканях (см. далее и гл. 284). Гемохроматоз означает прогрессирующую и массивную перегрузку тканей железом, что приводит к фиброзу и недостаточности органов. Несмотря на то что в отношении дефиниций имеются разногласия, логично, по-видимому, пользоваться следующей терминологией: 1) «генетический гемохроматоз» — врожденное заболевание, о котором в настоящее время известно, что оно связано с патологией гена насыщения тканей железом; этот ген прочно сцеплен с А-локусом комплекса HLA на 6-й хромосоме; 2) «приобретенный гемохроматоз» — перегрузка железом с повреждением тканей, вторичная по отношению к другому заболеванию, обычно талассемии или сидеробластической анемии. Следует подчеркнуть, однако, что при приобретенных заболеваниях, сопровождающихся отложениями железа, его большие скопления в паренхиматозных тканях могут обусловить те же клинические и патоморфологические последствия, что и генетический гемосидероз.
Метаболический дефект, обусловливающий повышенное всасывание железа при гемохроматозе, неизвестен. В настоящее время генетическое заболевание можно диагностировать на его ранних стадиях, когда отложения железа еще невелики, а повреждение органов минимально. На этой стадии болезнь предпочтительнее называть латентным или прецирротическим гемохроматозом (рис. 310-1).
Распространенность. Генетический гемохроматоз — не столь редкое заболевание, как полагали ранее. Среди представителей европеоидной популяции распространенность гена этой болезни составляет примерно 5 %, сама болезнь (гомозиготы) встречается с частотой около 0,3 %, а носительство (гетерозиготы) — 10 %. Вместе с тем на экспрессию болезни влияет ряд факторов, особенно кровопотеря, связанная у женщин с менструациями и родами. У мужчин это заболевание встречается в 5—10 раз чаще, чем у женщин. Почти у 70 % больных первые симптомы появляются в возрасте 40—60 лет. В возрасте до 20 лет клинические проявления болезни отмечаются редко, хотя при семейном скрининге (см. далее) могут быть выявлены не предъявляющие жалоб лица с избыточным содержанием железа, в том числе молодые менструирующие женщины.
Патогенез. В норме количество железа в организме (3—4 г) поддерживается за счет того, что его всасывание слизистой оболочкой кишечника соответствует его потерям. Эти величины составляют примерно 1 мг/сут у мужчин и 1,5 мг/сут у менструирующих женщин. При гемохроматозе всасывание железа слизистой оболочкой кишечника превышает потребности организма, достигая 4 мг/сут и более.; Накопление вследствие этого железа проявляется вначале повышением его уровня в плазме и увеличением насыщенности трансферрина. По мере прогрессирования болезни содержание железа в организме может достигать более 20 г. Этот избыток откладывается главным образом в паренхиматозных клетках печени, поджелудочной железы и сердца. Количество железа в печени и поджелудочной железе увеличивается в 50—100 раз, в сердце — в 5—25 раз, в селезенке, почках и коже — примерно в 5 раз. Повреждение тканей может быть связано с разрывом перегруженных железом лизосом и перекисным окислением липидов субклеточных органелл под влиянием избытка железа. Обнаружение связи между гемохроматозом и антигенами комплекса гистосовместимости HLA-A3, HLA-B14 и HLA-B7 подтвердило генетическую основу болезни. Она наследуется как аутосомный рецессивный признак, и у гомозигот обычно развивается тяжелая перегрузка железом с сопутствующими симптомами, а у гетерозигот незначительно нарушается обмен железа без прогрессирующих его отложений или клинических проявлений болезни. Отложение больших количеств железа в паренхиматозных органах, т. е. приобретенный гемохроматоз, встречается при хронических нарушениях эритропоэза, особенно при нарушении синтеза гемоглобина и неэффективном эритропоэзе, характерном для сидеробластической анемии и талассемии. При болезнях этой группы всасывание железа усилено, кроме того, больных часто лечат препаратами железа и гемотрансфузиями. Избыточное отложение железа в паренхиматозных органах иногда происходит и при медленно развивающейся кожной порфирии (porphyria cutanea tarda) — заболевании, характеризующемся нарушением биосинтеза порфирина (см. гл. 312). Однако степень перегрузки железом при этом обычно не столь велика, чтобы вызвать повреждение тканей.
У больных алкоголизмом с хронической патологией печени могут увеличиваться запасы железа в тканях. Этих больных можно разделить на две группы. У одних незначительно или умеренно повышается уровень окрашиваемого железа в печени, но общее его количество в организме остается относительно нормальным. Они страдают алкогольным повреждением печени (обычно цирроз), но не гемохроматозом. Причина увеличения запасов железа в печени может заключаться отчасти в некрозе клеток и поглощении высвобождаемого железа соседними купферовскими и паренхиматозными клетками. У больных второй (меньшей по численности) группы, страдающих алкоголизмом, повышен уровень железа в печени и выявляются его большие скопления и увеличение общего количества в организме. Обычно у них можно выявить генетический гемохроматоз с сопутствующим алкогольным повреждением печени или без него. Гемохроматоз у многих лиц, злоупотребляющих алкоголем, можно отдифференцировать от алкогольного повреждения печени, двумя методами: 1) определяя концентрацию железа в печени (см. далее и табл. 310-1) и 2) обследуя родственников на предмет выявления признаков заболевания, включая HLA-типирование членов семьи.
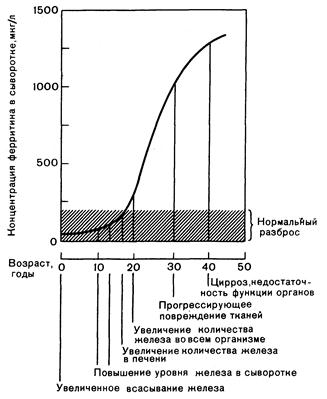
Рис. 310-1. Последовательность изменений при врожденном гемохроматозе и их корреляция с уровнем ферритина в сыворотке. Увеличенное всасывание железа происходит в течение всей жизни больного. Явное, сопровождающееся клиническими симптомами заболевания развивается обычно в возрасте 40—60 лет, но латентная предсклеротическая стадия болезни может быть диагностирована значительно раньше.
Сообщалось, что избыточное потребление железа в течение многих лет приводит к появлению клинических и патоморфологических признаков гемохроматоза. Это было отмечено, например, у некоторых представителей негроидной популяции, проживающих в Южной Африке (банту), у которых избыточное потребление железа с алкогольными напитками обусловлено способом приготовления последних в железных сосудах. Имеется несколько разрозненных сообщений о гемохроматозе, развившемся, по-видимому, у исходно здоровых лиц, в течение многих лет принимающих содержащие железо препараты, но не исключено, что дефект у них был наследственным.
Аномалия, лежащая в основе гемохроматоза, неизвестна. В связи с этим болезнь диагностируют по фенотипическим проявлениям (например, увеличение запасов железа в организме), которые могут зависеть от других факторов, таких как кровопотеря или потребление железа. Общей чертой у всех больных с гемохроматозом служит избыток железа в паренхиматозных органах. Парентеральное введение железа при трансфузиях его препаратов сопровождается перегрузкой железом преимущественно ретикулоэндотелиальных клеток. По-видимому, это сопровождается меньшим повреждением тканей, чем перегрузка железом клеток паренхимы.
Таблица 310-1. Уровень железа у здоровых, больных гемохроматозом и больных с алкогольным повреждением печени
| Показатель | Норма | Симптоматический гемохроматоз | Гомозиготы с ранним бессимптомным гемохроматозом | Алкогольное повреждение печени |
| Уровень железа в плазме, мкг/л | 500—1500 | 1800—3000 | Обычно повышен | Часто повышен |
| Общая железосвязывающая способность, мкг/л | 2500—3700 | 2000—3000 | 2000—3000 | 2500—3700 |
| Процент насыщенного трансферрина, мкг/л | 220—460 | 500—1000 | 500—1000 | 220—600 |
| Содержание трансферрина в сыворотке, нг/мл | 10—200 | 900—6000 | 200—500 | Ю—500 |
| Экскреция железа с мочой', мг/сут | 0—2 | 9—23 | 2—5 | Обычно менее 5 |
| Содержание железа в печени, мкг/100 мг сухой массы | 30—140 | 600—1800 | 200—400 | 30—200 |
' После внутримышечного введения 0,5 г деферроксамина.
Патоморфология. При аутопсии можно видеть увеличенные бугристые печень и поджелудочную железу необычного желто-коричневого цвета. Во многих органах, особенно в печени и поджелудочной железе и в меньшей степени в эндокринных железах и сердце, гистологически определяется железо. Исключение составляют яички, в которых количество железа относительно невелико, несмотря на то что их недостаточность относится к типичным и ранним признакам болезни. В отличие от этого гипофиз почти всегда вовлекается в процесс. Эпидермис бывает истончен, в клетках базального слоя увеличено количество меланина. Отложения железа окружают клетки, выстилающие синовиальную оболочку суставов, в которых видны кристаллы пирофосфата кальция, погруженные в синовиальную ткань.
Железо, откладывающееся в паренхиме печени у больных с генетическим гемохроматозом, находится в форме ферритина и гемосидерина. На ранних стадиях болезни его отложения обнаруживаются в перипортальных клетках паренхимы, особенно внутри лизосом на периферии цитоплазмы гепатоцитов. Эта стадия прогрессирует до фиброза долек и отложения железа в эпителии желчных протоков, купферовских клетках и фиброзных перегородках. Воспалительных клеток мало, но выражена пролиферация мелких желчных протоков. В ткани клиновидных биоптатов виден характерный фиброз с плотными фиброзными перегородками, окружающими дольки, что несколько напоминает хроническое поражение желчных путей. В поздней стадии развивается макроузелковый или смешанный макро- и микроузелковый цирроз.
Клинические проявления. Симптомы и признаки гемохроматоза включают в себя пигментацию кожи, диабет, дисфункцию печени и сердца, артропатию и гипогонадизм. Ранние жалобы чаще всего сводятся к слабости, апатии, похуданию; изменению окраски кожи, болям в животе, утрате либидо и явлениям, связанным с началом диабета. Наиболее четкими объективными признаками полностью развившегося заболевания служат гепатомегалия, пигментация, паукообразные ангиомы, спленомегалия, артропатия, асцит, нарушения сердечного ритма, застойная сердечная недостаточность, выпадение волос на теле, атрофия яичек и желтуха.
Печень обычно первой вовлекается в процесс, и гепатомегалия выявляется более чем у 95 % предъявляющих жалобы больных. Печень может быть увеличена и при отсутствии жалоб или при неизмененных функциональных печеночных пробах. Действительно, более чем у половины больных с симптомами гемохроматоза лабораторные признаки нарушения печеночных функций скудны или вообще отсутствуют, несмотря на гепатомегалию и фиброз. Часто отмечаются выпадение волос на теле, эритема ладонных поверхностей, атрофия яичек и гинекомастия. Портальная гипертензия и варикозное расширение вен пищевода встречаются реже, чем при циррозе Лаэннека. Примерно у половины предъявляющих жалобы больных увеличивается поджелудочная железа. Рак печени развивается почти у 30 % больных. Частота этого осложнения с возрастом увеличивается, и в настоящее время оно служит наиболее частой причиной смерти больных с гемохроматозом. Однако рак развивается, по-видимому, только при циррозе, что подчеркивает важность его ранней диагностики и начала лечения.
Чрезмерная пигментация кожи определяется почти в 90 % случаев к моменту установления диагноза. Отложения меланина в коже обычно способствует загару. Характерный металлически-серый цвет также объясняют увеличением содержания в коже меланина или меланина вместе с железом. Пигментация обычно диффузна и захватывает всю кожную поверхность тела, но часто она особенно выражена на лице, шее, разгибательной поверхности нижней части предплечий, тыльной стороне кистей, нижних отделах голеней, в области половых органов и кожных рубцов. Пигментацию слизистой оболочки рта удается обнаружить только в 10—15 % случаев. Иногда пигментируются твердое небо и сетчатка.
Сахарный диабет развивается примерно у 65 % больных и с большей вероятностью при диабете в семейном анамнезе. В патогенезе диабета при гемохроматозе принимают участие, таким образом, оба фактора, т. е. наследственная отягощенность и непосредственное повреждение отложениями железа поджелудочной железы. Течение диабета в этих случаях не отличается от такового при идиопатическом сахарном диабете, за исключением более частой инсулинорезистентности и липодистрофии. Поздние дегенеративные осложнения также не отличаются от последствий сахарного диабета.
Артропатия развивается у 25—50 % больных. Чаще всего она отмечается у лиц в возрасте старше 50 лет, но может присоединиться в любой стадии болезни и иногда бывает ее первым проявлением или появляется значительно позднее начала лечения. Вначале, как правило, в процесс вовлекаются мелкие суставы рук, особенно II и III пястно-фаланговые суставы. Прогрессирующий полиартрит может распространяться на лучезапястные, бедренные и коленные суставы. Могут появляться острые кратковременные приступы синовита, связанные с отложением пирофосфата кальция (хондрокальциноз, или псевдоподагра), главным образом в коленных суставах. Рентгенологически выявляются кистозные изменения склерозированных субхондральных поверхностей костей, утрата суставных хрящей с сужением суставных пространств, диффузная деминерализация, гипертрофическая пролиферация кости и кальциноз синовиальных оболочек. Механизм этих нарушений и их связь с обменом железа неизвестны.
Патология сердца определяется почти у 15 % больных. Наиболее частым их проявлением служит застойная сердечная недостаточность: примерно у 10 % больных молодого возраста. Ее симптомы могут появиться внезапно, и без лечения она быстро заканчивается смертью больного. Сердце диффузно увеличивается, в результате чего можно ошибочно поставить диагноз идиопатической кардиомиопатии, если отсутствуют другие явные признаки гемохроматоза. Могут присоединиться разнообразные нарушения сердечного ритма, особенно суправентрикулярные экстрасистолы и пароксизмальные тахиаритмии. Иногда происходят мерцание предсердий, предсердные фибрилляции и атриовентрикулярная блокада разной степени.
Часто больной утрачивает либидо, и у него атрофируются яички. Утрата либидо может предшествовать другим клиническим проявлениям. Атрофия яичек обычно бывает обусловлена сниженной продукцией гонадотропинов в связи с нарушением гипоталамо-гипофизарной функции из-за отложений железа. Иногда больные страдают также недостаточностью надпочечников, гипотиреозом и гипопаратиреозом.
Диагностика. Сочетание гепатомегалии, пигментации кожи, сахарного диабета, патологии сердца, артрита и признаков гипогонадизма позволяет предположительно поставить диагноз гемохроматоза. Однако сравнительно кратковременная и умеренная перегрузка железом паренхиматозных органов может вообще не сопровождаться клинической симптоматикой или проявляется лишь некоторыми из них, например, у лиц молодого возраста (см. рис. 310-1). В связи с этим предположить гемохроматоз следует каждый раз при необъяснимой гепатомегалии, идиопатической кардиомиопатии, необычной пигментации кожи, утрате либидо, диабете или артрите.
Анамнез следует особенно детализировать в отношении заболевания других членов семьи, потребления алкоголя, приема железа и больших доз аскорбиновой кислоты, способствующей его всасыванию. При анализе крови обращают внимание на выраженность анемии и нарушения эритропоэза с целью исключить перегрузку железом на почве гематологических расстройств. Повреждение печени, поджелудочной железы, сердца и суставов должно быть подтверждено осмотром, данными рентгенологического исследования и результатами функциональных проб. Затем выявляют общее увеличение запасов железа в организме и особенно увеличение его количества в паренхиматозных органах, сопровождающееся повреждением тканей.
Существующие методы выявления избытка железа включают: 1) определение его уровня в сыворотке; 2) определение процента насыщения трансферрина; 3) оценку запасов хелируемого железа с помощью препарата дефероксамина; 4) определение концентрации ферритина в сыворотке; 5) биопсию печени (см. табл. 310-1) и 6) компьютерную томографию. Каждый из этих методов обладает своими преимуществами и недостатками. Содержание железа в сыворотке и процент насыщения трансферрина увеличиваются уже на ранних стадиях болезни, но специфичность этих показателей ограничивается сравнительно большим числом ложноположительных и ложноотрицательных результатов. В частности, количество железа в сыворотке может увеличиваться при алкогольном повреждении печени без перегрузки железом; при этом, однако, в отличие от гемохроматоза не снижается железосвязывающая способность сыворотки (см. табл. 310-1).
Содержание ферритина в сыворотке обычно представляет собой надежный показатель запасов железа в организме независимо от того, уменьшены они или увеличены. У нелеченых больных с гемохроматозом уровень ферритина в сыворотке резко повышен (см. рис. 310-1 и табл. 310-1). Определение ферритина служит также неинвазивным тестом при скрининге в ранние стадии болезни, так как они изменяются обычно еще до появления каких-либо морфологических признаков повреждения печени, а концентрация ферритина коррелирует с запасами железа в организме. В связи с этим тест и применяют обычно вместо более громоздких методов скрининга, включающих в себя определение экскреции железа с мочой. Однако у больных с воспалительными процессами и некрозом печеночных клеток уровень ферритина в сыворотке может повышаться в несоответствии с запасами железа в организме. Выделено также несколько семей, у больных членов которых уровень ферритина в сыворотке оставался в пределах нормы, несмотря на увеличение запасов железа. Причина этого расхождения неясна, но, по-видимому, оно представляет собой исключение из правила. В клинической практике сочетанное определение концентрации железа в сыворотке, процента насыщения трансферрина и уровня ферритина в сыворотке формирует наиболее простой и наиболее надежный скрининг-тест на гемохроматоз, в том числе в прецирротическую стадию болезни. При отклонении от нормы результатов любого из этих тестов следует производить биопсию печени, так как именно она служит решающей для подтверждения диагноза гемохроматоза. При этом можно гистохимически определить железо в ткани, измерить его печеночную концентрацию и выяснить степень повреждения ткани. При компьютерной томографии находят уплотнение печени вследствие отложения в ней железа. Однако для этого необходимы специальное оборудование и опытный персонал, кроме того, неясны нижние границы увеличенного количества железа в тканях, что затрудняет точную оценку.
При установлении диагноза гемохроматоза принципиально важно обследовать членов семьи больного. У больных членов семьи независимо от жалоб уровень железа в плазме обычно повышен, общая железосвязывающая способность сыворотки снижена, насыщение трансферрина увеличено, концентрация ферритина в сыворотке увеличена или увеличивается. Эти изменения регистрируются даже еще до выраженного увеличения запасов железа в организме (см. рис. 310-1). Затем следует произвести биопсию печени, поскольку она совершенно необходима для установления диагноза и начала лечения до развития повреждения тканей. При обследовании членов семей, относящихся к группе риска, полезно проводить HLA-типирование. Больные сиблинги обычно обладают идентичными с пробандом гаплотипами HLA, а при заболеваании детей пробанда следует предполагать гомозиготно-гетерозиготное скрещивание.
Различия между гемохроматозом и алкогольным циррозом, сопровождающимся повышением тканевого уровня железа, уже обсуждались. Дифференцировать эти состояния обычно нетрудно, если определить концентрацию железа в печени, а тест на экскрецию железа после введения деферроксамина может обеспечить дополнительной информацией (см. табл. 310-1).
Лечение. При генетическом гемохроматозе лечение предусматривает выведение избытка железа из организма и поддержание функции поврежденных органов.
Железо легче всего выводится из организма при еженедельном однократном (или один раз в 2 нед) кровопускании в объеме 500 мл. Несмотря на то что при этом вначале несколько уменьшается объем эритроцитной массы (примерно до 350 мл/л), через несколько недель он стабилизируется. Уровень железа в плазме остается повышенным до истощения его запасов. Уровень ферритина в плазме прогрессивно снижается, что отражает постепенное уменьшение запасов железа в организме. Поскольку в 500 мл крови содержится 200—250 мг железа, а удалять его следует в количестве примерно 25 г, обычно еженедельные кровопускания проводят в течение 2—3 лет. После нормализации уровней железа и ферритина в плазме кровопускания производят с такими интервалами, чтобы обеспечить поддержание железа в плазме на уровне ниже 1500 мкг/л. Обычно достаточно производить по одной флеботомии каждые 3 мес. Адекватность лечения можно оценить в любое время, определив уровень железа в плазме, процент насыщения железом трансферрина или концентрацию ферритина в сыворотке. Эти показатели быстро достигают патологически высокого уровня при реаккумуляции железа.
|
|
|
|
|
Дата добавления: 2014-11-20; Просмотров: 454; Нарушение авторских прав?; Мы поможем в написании вашей работы!