КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Нарушение транспорта анионок хлоридорея 4 страница
|
|
|
|
При парентеральном введении хелирующих агентов, таких как дефероксамин, за сутки выводится 10—20 мг железа, т. е. меньше половины того количества, которое мобилизуется еженедельным кровопусканием. Кроме того, флеботомия при лечении больных с генетическим гемохроматозом, как правило, обходится дешевле, более удобна и безопасна. Однако хелирующие агенты показаны в тех случаях, когда анемия или гипопротеинемия служит препятствием для кровопускания. Наиболее эффективны подкожные инфузии деферроксамина с помощью портативного насоса, работающего на малой скорости.
Лечение при печеночной и сердечной недостаточности и диабете мало отличается от обычного, проводимого при этих болезнях. Утрату либидо и изменение вторичных половых признаков частично компенсируют тестостероном или гонадотропинами.
Прогноз. Основными причинами смерти нелеченых больных служат сердечная (30 %) и печеночная недостаточность или портальная гипертензия (25 %) и гепатоклеточная карцинома (30 %).
Путем выведения избытка железа и поддержания его на уровне, близком к норме, ожидаемую продолжительность жизни у больных с клиническими признаками болезни удается увеличить более чем на 8 лет. Лечение увеличивает показатель 5-летней выживаемости с 33 до 89 %. При выведении железа с помощью повторных кровопусканий печень и селезенка уменьшаются в размерах, показатели печеночных функций нормализуются, пигментация кожи уменьшается, а сердечная недостаточность корригируется. Толерантность к углеводам повышается примерно у 40 % больных. Однако на гипогонадизм или артропатию удаление избытка железа практически не влияет. Фиброз печени может уменьшаться, но цирроз необратим. Несмотря на адекватное выведение железа из организма, примерно у 1/3 больных на поздних стадиях развивается гепатоклеточная карцинома. Кажущееся увеличение ее частоты у леченых больных объясняется, вероятно, увеличением продолжительности их жизни. Это осложнение, по-видимому, не развивается, если лечение начато в прецирротической стадии болезни, поэтому невозможно переоценить значение обследования членов семьи и раннего начала лечения. Больным без клинических симптомов, выявленным при семейном скрининге, следует проводить кровопускания, если запасы железа у них значительно или даже умеренно увеличены. Важно также периодически оценивать динамику этих запасов. В этом случае удается предотвратить большинство проявлений- болезни. Действительно, ожидаемая продолжительность жизни у больных, лечение которых начато еще в прецирротической стадии, очевидно, не слишком отличается от таковой в здоровой популяции.
ГЛАВА 311. БОЛЕЗНЬ ВИЛЬСОНА
Я. Герберт Шайнберг (I. Herbert Schein berg)
Болезнь Вильсона представляет собой наследуемое по аутосомному рецессивному типу нарушение печеночной экскреции меди, сопровождающееся накоплением ее токсических количеств в печени, мозге и других органах. Заболевают лица любой этнической и географической принадлежности, общая распространенность болезни составляет примерно 1:30 000. Ее характерной особенностью служит недостаточность в плазме содержащего медь белка церулоплазмина.
Естественное развитие. У здоровых новорожденных количество церулоплазмина в плазме невелико, а уровень железа в печени высок. В течение первого года жизни уровень церулоплазмина повышается, а концентрация меди в печени уменьшается, достигая уровня, типичного для взрослого человека. В отличие от этого у гомозигот по гену болезни Вильсона содержание церулоплазмина в плазме изменяется незначительно, и концентрация меди в печени остается повышенной. Однако клинические признаки избытка меди редко появляются в возрасте до 6 лет, а у половины нелеченых больных симптомы отсутствуют и в подростковом возрасте.
Примерно в половине случаев болезнь Вильсона проявляется печеночной патологией. Токсические эффекты меди в печени обнаруживают себя острым гепатитом, циррозом или бессимптомной гепатоспленомегалией. Острый гепатит напоминает вирусный, его можно спутать с инфекционным мононуклеозом, и он может развиваться в трех разных направлениях. Первое — это скоротечная, иногда летальная форма болезни, проявляющаяся желтухой, недомоганием и подчас асцитом, гипоальбуминемией и повышением уровня печеночных ферментов в плазме. В острой фазе в плазму может поступать достаточное для индукции гемолитической анемии количество меди. Диагноз иногда не удается поставить до аутопсии или до того момента, когда заболевание младших сиблингов заставит произвести анализ сохранившихся тканей на медь. Второе направление — это постепенное повреждение паренхимы печени, обусловливающее клинические и гистологические изменения, неотличимые от таковых при хроническом активном гепатите. Третье — внешнее выздоровление больного после гепатита, несмотря на развитие в дальнейшем цирроза печени. До появления признаков или симптомов болезни может пройти несколько лет и даже десятилетий. В этих случаях без тщательного опроса больного или при отсутствии клинически выраженного цирроза можно и не обратить внимания на имевший место в прошлом приступ гепатита.
Чаще вызываемое скоплением меди повреждение печени переходит в цирроз без каких бы то ни было проявлений гепатита. В этом случае начальные проявления болезни бывают внепеченочными. У большинства больных к первым клиническим признакам относится неврологическая или психическая патология, что всегда сопровождается появлением колец Кайзера—Флейшера. Эти зеленого или золотистого цвета отложения меди в десцеметовой оболочке роговицы не мешают зрению, но указывают на высвобождение меди из печени и повреждение мозга. Иногда появлению колец сопутствуют подсолнечниковые катаракты. Если у больного с выраженной неврологической или психической патологией опытный врач при обследовании с помощью щелевой лампы не находит колец Кайзера—Флейшера, диагноз болезни Вильсона можно исключить.
Первыми неврологическими проявлениями служат нарушения двигательных функций, особенно тремор в покое и при произвольных движениях. Часты спастичность и ригидность мышц, хорея, навязчивые движения, дисфагия и дизартрия. Иногда определяется рефлекс Бабинского и отсутствуют брюшные рефлексы. Чувствительность не нарушается. У большинства больных клиническая симптоматика сопровождается нарушениями психики, что обусловлено отчасти токсическими эффектами меди в головном мозге, а отчасти осознанием смертельно опасной болезни. Могут появляться синдромы, неотличимые от шизофрении, маниакально-депрессивных психозов и классических неврозов, а некоторые странности поведения затрудняют классификацию этих состояний. При уменьшении избытка меди с помощью фармакологических средств психическое состояние больного может улучшиться, но часто требуется и психотерапия.
В отдельных случаях болезнь начинается не с дисфункций печени или ЦНС. Например, у некоторых молодых женщин ее первым признаком может быть первичная или вторичная аменорея, другие страдают повторными спонтанными выкидышами из-за избытка свободной меди в маточном секрете. Иногда диагностике помогает обычное офтальмологическое обследование больного, при котором обнаруживают кольца Кайзера—Флейшера в отсутствие печеночных или неврологических симптомов.
Патогенез. Метаболические нарушения заключаются в невозможности поддержания почти нулевого баланса меди в организме. Накопление меди происходит, вероятно, потому, что печеночные лизосомы утрачивают нормальный механизм ее экскреции в желчь, поступающей в печень при отщеплении от разрушающегося церулоплазмина. Это может обусловливать дефицит церулоплазмина, поскольку стехиометрический избыток меди ингибирует его образование из апоцерулоплазмина и меди. В конце концов способность гепатоцитов накапливать медь оказывается превышенной, и металл высвобождается в кровь и поглощается внепеченочными тканями (табл. 311-1).
В норме практически вся тканевая медь находится в простетических группах медьсодержащих ферментов, таких как цитохромоксидаза, тирозиназа, супероксиддисмутаза и церулоплазмин. Свободной (не связанной с белком) меди в норме мало или совсем нет. При болезни Вильсона в организме количество меди больше, чем способно связаться специфическими медьсодержащими белками. Она столь же токсична, как и находящиеся в избытке железо, цинк, ртуть или свинец. Токсичность этих катионов в большей степени объясняется, вероятно, их патологическим связыванием белками, в норме не содержащими металлов.
Патологические -скопления меди проявляются прежде всего патологией печени. При световой микроскопии в ней прежде всего выявляют чрезмерные скопления жира и гликогена (рис. 311-1), при электронной микроскопии—изменения митохондрий, специфичные для болезни Вильсона (рис. 311-2). Позднее появляются некроз, воспаление, фиброз, пролиферация желчных протоков и цирроз. Аномалии химизма печени присоединяются позднее.
Смерть может наступить в результате отравления медью центральной нервной системы и на фоне небольших признаков нарушения функции печени или вообще без них, но, как правило, раньше или позднее выявляется ее выраженное повреждение. Если болезнь продолжается достаточно долго, то цирроз печени развивается всегда.
Таблица 311-1. Результаты определения количества меди при болезни Вильсона, у гетерозиготных носителей и здоровых лиц
| Группа | Уровень церулоплазмина в сыворотке | Концентрация меди в печени | ||||
| число обследованных | колебания, мг/л | средняя^ стандартное отклонение, мг/л | число обследованных | колебания, мкг/г сухой массы | средняя ± стандартное отклонение, мкг/г сухой массы | |
| Болезнь Вильсона | ||||||
| Бессимптомное течение | 0—195 | 36±53 | 152—1828 | 983,5±3б8 | ||
| На фоне симптомов | 0—430 | 59±71 | 94—1360 | 5 8 8,3 ±304 | ||
| Гетерозиготные носители | 95' | 10—501 | 284±85 | 39—213 | 117,0±51 | |
| Здоровые лица | 185—659 | 307 ±35 | 20—45 | 31,5±б,8 |
' Из них 71 родитель больных детей и 24 ребенка, у которых один из родителей был болен.
Источник: Sternlieb a. Scheinberg, 1968.
В мозге избыток меди определяют постоянно. Некрозу нейронов с образованием полостей может предшествовать появление клеток Опальского или Альцгеймера II типа, однако они неспецифичны для болезни Вильсона.
В почках избыток меди практически не вызывает структурных изменений и обычно не сопровождается нарушением их функции. Гематурия, протеинурия, синдром Фанкони и канальцевый ацидоз определяются редко. Изменения других органов и тканей незначительны.
Диагностика. Диагноз не вызывает затруднений, если о нем помнить. Болезнь Вильсона следует подозревать у любого больного в возрасте до 40 лет с необъяснимыми дисфункцией ЦНС, признаками или симптомами хронического активного гепатита, повышением уровня сывороточной трансаминазы, гемолитической анемией на фоне гепатита или необъяснимым циррозом, а также у любого больного, родственники которого страдают болезнью Вильсона.
В сомнительных случаях диагноз подтверждают, если: 1) концентрация церулоплазмина в сыворотке составляет менее 200 мг/л в присутствии колец Кайзера—Флейшера либо 2) его концентрация менее 200 мг/л при содержании меди в биоптате печени более 250 мкг/г сухой массы. У большинства больных экскреция меди с мочой превышает 100 мкг/сут, а в биоптате печени определяются гистологические изменения.
Примерно у 5 % больных концентрация церулоплазмина в сыворотке превышает 200 мг/л, а избыток меди в печени и кольца Кайзера—Флейшера выявляют иногда и при другой патологии печени. Во всех этих случаях полезным дифференциально-диагностическим тестом служит определение способности включать радиоактивную медь в церулоплазмин. Даже при нормальной концентрации церулоплазмина при болезни Вильсона изотоп практически не включается в белок, тогда как при другой патологии печени и избытке в ней меди его включение не нарушается.
Лечение. Лечение направлено на возможно быстрое выведение избытка меди и должно начинаться сразу же после установления диагноза независимо от того, предъявляет больной жалобы или не предъявляет. Средством выбора служит D-пеницилламин. Его назначают для приема внутрь в начальной дозе 1 г/сут обычно в 2 приема: до еды и перед сном. Поскольку у животных пеницилламин оказывает антипиридоксиновое действие, больной должен одновременно получать по 25 мг/сут витамина Вв. Эффективность лечения оценивают химическими и клиническими методами. Вначале суточная экскреция меди с мочой должна увеличиться в 5 раз и более по сравнению с исходным уровнем, и в первый месяц лечения она может выводиться в количестве 1—3 мг/сут.
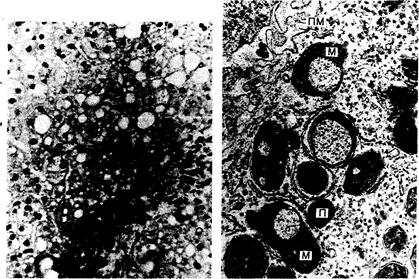
Рис. 311-1. Изменения жировых включений, депонирование гликогена и клеточные инфильтраты в окрашенном гематоксилином и эозином препарате печени мальчика с бессимптомной болезнью Вильсона.
Рис. 311-2. Электронная микрофотограмма биоптата печени 6-летнего мальчика с бессимптомной болезнью Вильсона.
В митохондриях (М) видны крупные вакуоли. П — пероксидаза, ПМ — плазматическая мембрана.
Несколько раз в неделю в первый месяц лечения и периодически в последующие периоды необходимо определять число лейкоцитов и тромбоцитов в крови, производить анализ мочи и регистрировать температуру тела. Обычно в первые 14 дней лечения появляются побочные реакции на пеницилламин: сыпь, лихорадочное состояние, лейко- и тромбоцитопения, лимфаденопатия или протеинурия. В этих случаях лечение следует прекратить. Его возобновляют с введения препарата в меньших, но постепенно увеличивающихся дозах. Побочные реакции менее вероятны, если в течение первых 2 нед лечения пеницилламином вводить 20 мг/сут преднизолона с его последующей постепенной отменой. Прежде чем удастся вводить пеницилламин без преднизолона, могут неоднократно развиваться реакции, при которых требуется введение десенсибилизирующих препаратов.
Лечение продолжают в течение всей жизни больного. Неадекватное лечение или перерывы в нем сопровождаются рецидивами, которые могут оказаться необратимыми. Возобновление приема пеницилламина после временного перерыва в лечении может вызвать появление или возобновление побочных реакций. В любое время, даже после многих лет отсутствия осложнений лечения, следует быть готовым к развитию гранулоцитопении (или агранулоцитоза), тромбоцитопении, нефротического синдрома, синдрома Гудпасчера, системной красной волчанки, тяжелых артралгий или злокачественной миастении. Токсичность иногда зависит от дозы, поэтому можно уменьшать дозу до терапевтически эффективной, но не токсичной. Непрерывное введение малых стероидов может предотвратить связанное с приемом пеницилламина развитие волчанки или артралгий. При нефротическом синдроме временная отмена препарата иногда позволяет возобновить лечение без рецидива протеинурии. Несмотря на то что больные с непреодолимой непереносимостью D-пеницилламина встречаются редко, его токсичность может быть столь выраженной, что требуется навсегда отказаться от лечения им. Введение в течение всей жизни больного димеркапрола вряд ли практически возможно, поэтому единственным альтернативным препаратом остается триентин.
В случае успешного начала лечения пеницилламином больного обследуют с 1—3-месячными интервалами в течение всей его жизни, чтобы не пропустить признаков токсичности препарата и следить за течением процесса. Более адекватными показателями эффективности лечения служат результаты врачебного обследования, в том числе надежной оценки неврологического статуса и осмотра роговицы с помощью щелевой лампы, а также собственное мнение больного о своем состоянии. Для характеристики состояния печеночной функции полезно проводить серийные определения уровней трансаминазы, альбумина и билирубина в сыворотке. Отсутствие клинического улучшения или ухудшение состояния может быть результатом развития необратимых повреждений еще до начала лечения, несоблюдения больным его режима или недостаточности дозы пеницилламина. Выяснению конкретной причины способствуют количественные определения экскреции меди с мочой и уровня свободной меди в сыворотке (общее ее количество, за исключением связанной с церулоплазмином). После длительного лечения экскреция меди с мочой должна быть ниже, чем в начале лечения, редко превышая 1,5 мг/сут. Еще более надежным показателем правильного лечения служит концентрация свободной меди в сыворотке обычно менее 100 мкг/л. Если в течение ряда лет больной не предъявляет жалоб, а лабораторные признаки нарушения функции печени отсутствуют, а также в случаях сохранения минимальных проявлений болезни без динамики, дозу пеницилламина можно уменьшить до 0,75 г/сут.
Лечение более 100 бессимптомных больных с подтвержденным диагнозом показало, что непрерывное введение D-пеницилламина может предотвращать практически все проявления болезни.
ГЛАВА 312. ПОРФИРИИ
Урс А. Мейер (Urs А. Меуег)
Порфирии представляют собой патологию, связанную с наследственными или приобретенными аномалиями биосинтеза гема. Порфирины, эти тетрапирроловые пигменты, выполняют роль промежуточных продуктов этого пути и образуются из предшественников — d-аминолевулиновой кислоты (АЛК) и порфобилиногена. Гем, комплекс двухвалентного железа с протопорфирином IX, функционирует в качестве простетической группы гемопротеинов, таких как гемоглобин, цитохромы, каталаза и триптофаноксигеназа. Его биосинтез жизненно важен и происходит во всех аэробных клетках.
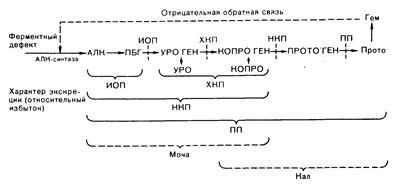
Рис. 312-1. Экскреция порфиринов и их предшественников с мочой при печеночных порфириях в связи с дефицитом ферментов биосинтеза гема. Интермедиаты этого пути, экскретируемые в избыточных количествах в острой фазе при любой печеночной порфирии, объединены фигурными скобками. АЛК — d-аминолевулиновая кислота, ПБГ — порфобилиноген, УРО'ГЕН — уропорфириноген, КОПРО'ГЕН — копропорфириноген, ПРОТО'ГЕН — протопорфириноген, ПРОТО — протопорфирин, ИОП—интермиттирующая острая порфирия, ХКП—хроническая кожная порфирия, НКП — наследственная копропорфирия, ПП — пестрая порфирия.
Каждая порфирия характеризуется особенностями гиперпродукции, накопления и экскреции промежуточных продуктов биосинтеза гема. Эти особенности отражают метаболическую экспрессию дефицита отдельных ферментов биосинтеза (табл. 312-1, рис. 312-1).
Основные клинические проявления заключаются в перемежающихся приступах дисфункции нервной системы и/или чувствительности кожи к солнечным лучам. Неврологический синдром обычно провоцируется приемом таких препаратов, как барбитураты, и заключается в болях в животе, периферической нейропатии и нарушениях психики. Нейропсихические симптомы появляются только при порфириях, при которых резко усилена продукция предшественников порфиринов — АЛК и порфобилиногена. Патогенез неврологических нарушений неясен. Светочувствительность кожи связана непосредственно с повышенным накоплением порфиринов, хотя при разных нарушениях кожные проявления неодинаковы. Светочувствительность обусловлена фотодинамическим действием порфиринов и опосредуется, вероятно, образующимся синглет-кислородом с последующим развитием деструктивных процессов, таких как перекисное окисление липидов лизосомных мембран. Доминантно наследуемые порфирии человека экспрессируются по-разному. Могут определяться лишь биохимические или ферментативные изменения. Подобное латентное течение болезни может оказаться одной из стадий или продолжается в течение всей жизни больного. В 'других случаях симптоматика может провоцироваться лекарственными препаратами, гормонами или повреждением печени.
Классификация. Порфирии подразделяют обычно на две основные группы (эритропоэтическая и печеночная; см. табл. 312-1) в соответствии с основными местами синтеза гема, в которых проявляются «ошибки» метаболизма. Единственная чисто эритропоэтическая форма порфирии — врожденная эритропоэтическая (ВЭП) — встречается редко. При протопорфирии (ПрП) порфирины накапливаются как в клетках эритроидного ряда, так и в печеночной ткани. При интермиттирующей острой порфирии, наследственной копропорфирии и пестрой порфирии (соответственно ИОП, НКП и ПП) доминантно наследуемая недостаточность ферментов обусловливает нарушение биосинтеза гема преимущественно в печени без видимых нарушений образования гемоглобина. Хроническую кожную порфирию (ХКП) ранее считали приобретенной печеночной. Однако у большинства (если не у всех) больных обнаруживают наследственную недостаточность уропорфириногендекарбоксилазы. Приобретенную порфирию, напоминающую ХКП, обусловливают воздействие полихлорированных углеводородов и опухоли печени. Отравление свинцом также сопровождается нарушением синтеза порфиринов и гема (см. гл. 172). Некоторое усиление экскреции порфиринов или их предшественников с мочой, равно как и накопление порфиринов в эритроцитах, может сопровождать многие клинические состояния. При вторичных феноменах симптомы и признаки порфирии отсутствуют.
Таблица 312-1. Характеристики порфирий
| Показатель | Порфирия | |||||
| эритропоэтическая врожденная | Печеночные | эритропече-ночная | ||||
| интермиттирующая острая | наследственная копропорфирия | пестрая порфирия | хроническая кожная | |||
| Недостаточность фермента | Порфобилино-гендезаминаза и/или уропорфи-риноген-Ш-ко-синтаза (?) | Порфобилино-гендезаминаза | Копропорфири-ногеноксидаза | Протопорфири-ногеноксидаза | Уропорфирино-гендекарбокси-лаза | Феррохелатаза |
| Наследование | Аутосомное рецессивное | Аутосомное доминантное | Аутосомное доминантное | Аутосомное доминантное | Аутосомное доминантное | Аутосомное доминантное |
| Метаболическая экспрессия | Эритроидные клетки | Печень | Печень | Печень | Печень | Эритроидные клетки и печень |
| Признаки и симптомы | ||||||
| Светочувствительность | Да | Нет | Редко | Да | Да | Да |
| кожи | ||||||
| Приступы болей в животе, нейропсихиче-ский синдром | Нет | Да | Да | » | Нет | Нет |
| Лабораторные данные | ||||||
| Эритроциты | ||||||
| Уропорфирин | +++ | В пределах нормы | В пределах нормы | В пределах нормы | В пределах нормы | В пределах нормы |
| Копропорфирин | ++ | То же | То же | То же | То же | + |
| Протопорфирин | (+) | »» | »» | »» | »» | -1-4-4- |
| Моча | ||||||
| d-Аминолевулиновая кислота | В пределах нормы | (+++) | (+4-4-) | (+++) | »» | В пределах |
| Порфобилиноген | То же | (+++) | (+++) | (+++) | »» | То же |
| Уропорфирин | +++ | ++ | + | + | +++ | »» |
| Копропорфирин | ++ | В пределах | ++ | ++ | + | (+) |
| Кал | нормы | |||||
| Копропорфирин | + | То же | +++ | + | (+) | (4-) |
| Протопорфирин | + | »» | + | +++ | В пределах | 4-+ |
| нормы |
Примечание. +—экскреция усилена, ++—умеренно повышена, +++—резко повышена, (+)—усилена у некоторых
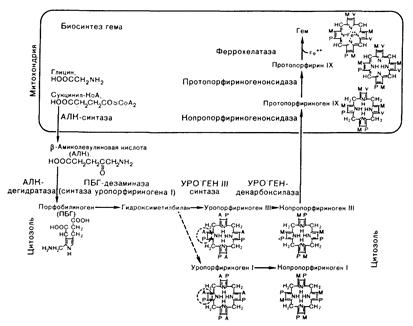
Рис. 312-2. Схема биосинтеза гема.
АЛК — d-аминолевулиновая кислота, ПБГ — порфобилиноген, УРО`ГЕН — уропорфириноген, А — уксусная кислота, Р — пропионовая кислота, М — метил, V — винил.
Биохимические аспекты. Последовательность реакций синтеза гема из субстратов глицина и сукцинил-фермента А через АЛК и порфобилиноген (ПБК) катализируется четырьмя митохондриальными и четырьмя цитозольными ферментами (рис. 312-2). В регуляции биосинтеза гема в разных тканях существуют различия.
В печени скорость образования гема ограничивается реакцией, катализируемой АЛК-синтазой. Ферменты, действующие после АЛК-синтазы, определяются в избытке. Главным регулятором АЛК-синтазы служит конечный продукт всего пути — гем, который репрессирует фермент по механизму отрицательной обратной связи. Повышенные потребности в геме удовлетворяются путем новообразования АЛК-синтазы. Ее синтез в печени индуцируется большим числом жирорастворимых веществ, стероидами и химическими соединениями, которые служат субстратами и индукторами гемопротеиновых цитохромов P450 — конечных оксидаз на пути микросомного метаболизма фармакологических средств. Эта индукция модулируется многочисленными генетическими, метаболическими и факторами окружающей среды. При порфириях, при которых симптомы провоцируются некоторыми лекарственными препаратами, взаимозависимость синтеза гема и микросомального окисления этих препаратов приобретает большое значение.
В клетках костного мозга, в которых происходит полный синтез гема, ограничивающая скорость реакция также катализируется АЛК-синтазой, но о ее роли в синтезе гема во время деления, дифференцировки и созревания клеток эритроидного ряда известно мало. В процессе созревания этих клеток из них исчезают ядра и митохондрии' и, следовательно, митохондриальные ферменты синтеза гема, тогда как цитозольные ферменты, катализирующие реакции между АЛ К и копропорфириногеном, сохраняются. В связи с этим эритроциты можно использовать для диагностики порфирий, связанных с дефектом только цитозольного фермента.
Регуляция синтеза гема в костном мозге и печени различна. В печени основной детерминантой образования гема служит уровень АЛК-синтазы, тогда как в костном мозге синтез гема запускается сложным процессом дифференцировки эритроидной клетки. Именно поэтому, вероятно, дефекты ферментов синтеза гема в эритроидных клетках и печени проявляются по-разному.
Порфириногены занимают промежуточное положение между порфобилиногеном и протопорфирином. Они бесцветны и не флюоресцируют. За исключением протопорфирина, порфирины — это побочные продукты, которые покидают путь биосинтеза вследствие необратимого окисления соответствующего порфириногена. Порфирины не выполняют физиологической функции, но в силу своей окраски и флюоресценции определяют необычный цвет мочи и эритроцитов у некоторых больных.
От расположения двух замещенных боковых цепей на пирроловом кольце порфиринов зависят структурные типы изомеров, которые нумеруются от I до IV. В природе найдены только типы I и III, причем только тип III служит субстратом конечных этапов реакции, ведущей к образованию протопорфирина IX и гема. При распаде гема образуются не порфирины, а нециклические тетрапирролы, называемые желчными пигментами.
|
|
|
|
|
Дата добавления: 2014-11-20; Просмотров: 414; Нарушение авторских прав?; Мы поможем в написании вашей работы!