КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Цепные реакции. 1 страница
|
|
|
|
Цепными называют реакции, состоящие из ряда взаимосвязанных стадий, когда частицы, образующиеся в результате каждой стадии, генерируют последующие стадии. Как правило, цепные реакции протекают с участием свободных радикалов. Для всех цепных реакций характерны три типичные стадии, которые мы рассмотрим на примере фотохимической реакции образования хлороводорода.
1. Зарождение цепи (инициация):
Сl2 + hν ––> 2 Сl•
2. Развитие цепи:
Н2 + Сl• ––> НСl + Н•
Н• + Сl2 ––> НСl + Сl•
Стадия развития цепи характеризуется числом молекул продукта реакции, приходящихся на одну активную частицу – длиной цепи.
3. Обрыв цепи (рекомбинация):
Н• + Н• ––> Н2
Сl• + Сl• ––> Сl2
Н• + Сl• ––> НСl
Обрыв цепи возможен также при взаимодействии активных частиц с материалом стенки сосуда, в котором проводится реакция, поэтому скорость цепных реакций может зависеть от материала и даже от формы реакционного сосуда.
Реакция образования хлороводорода является примером неразветвленной цепной реакции – реакции, в которой на одну прореагировавшую активную частицу приходится не более одной вновь возникающей. Разветвленными называют цепные реакции, в которых на каждую прореагировавшую активную частицу приходится более одной вновь возникающей, т.е. число активных частиц в ходе реакции постоянно возрастает. Примером разветвленной цепной реакции является реакция взаимодействия водорода с кислородом:
1. Инициация:
Н2 + О2 ––> Н2О + О•
2. Развитие цепи:
О• + Н2 ––> Н• + ОН•
Н• + О2 ––> О• + ОН•
ОН• + Н2 ––> Н2О + Н•
2.1.9 Влияние температуры на константу скорости реакции
Константа скорости реакции есть функция от температуры; повышение температуры, как правило, увеличивает константу скорости. Первая попытка учесть влияние температуры была сделана Вант-Гоффом, который сформулировал следующее эмпирическое правило:
При повышении температуры на каждые 10 градусов константа скорости элементарной химической реакции увеличивается в 2 – 4 раза.
Величина, показывающая, во сколько раз увеличивается константа скорости при повышении температуры на 10 градусов, есть температурный коэффициент константы скорости реакции γ. Математически правило Вант-Гоффа можно записать следующим образом:
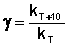 (II.29)
(II.29)
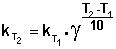 (II.30)
(II.30)
Однако правило Вант-Гоффа применимо лишь в узком температурном интервале, поскольку температурный коэффициент скорости реакции γ сам является функцией от температуры; при очень высоких и очень низких температурах γ становится равным единице (т.е. скорость химической реакции перестает зависеть от температуры).
2.1.10 Уравнение Аррениуса
Очевидно, что взаимодействие частиц осуществляется при их столкновениях; однако число столкновений молекул очень велико и, если бы каждое столкновение приводило к химическому взаимодействию частиц, все реакции протекали бы практически мгновенно. Аррениус постулировал, что столкновения молекул будут эффективны (т.е. будут приводить к реакции) только в том случае, если сталкивающиеся молекулы обладают некоторым запасом энергии – энергией активации.
Энергия активации есть минимальная энергия, которой должны обладать молекулы, чтобы их столкновение могло привести к химическому взаимодействию.
Рассмотрим путь некоторой элементарной реакции
А + В ––> С
Поскольку химическое взаимодействие частиц связано с разрывом старых химических связей и образованием новых, считается, что всякая элементарная реакция проходит через образование некоторого неустойчивого промежуточного соединения, называемого активированным комплексом:
А ––> K# ––> B
Образование активированного комплекса всегда требует затраты некоторого количества энергии, что вызвано, во-первых, отталкиванием электронных оболочек и атомных ядер при сближении частиц и, во-вторых, необходимостью построения определенной пространственной конфигурации атомов в активированном комплексе и перераспределения электронной плотности. Таким образом, по пути из начального состояния в конечное система должна преодолеть своего рода энергетический барьер. Энергия активации реакции приближённо равна превышению средней энергии активированного комплекса над средним уровнем энергии реагентов. Очевидно, что если прямая реакция является экзотермической, то энергия активации обратной реакции Е'А выше, нежели энергия активации прямой реакции EA. Энергии активации прямой и обратной реакции связаны друг с другом через изменение внутренней энергии в ходе реакции. Вышесказанное можно проиллюстрировать с помощью энергетической диаграммы химической реакции (рис. 2.5).
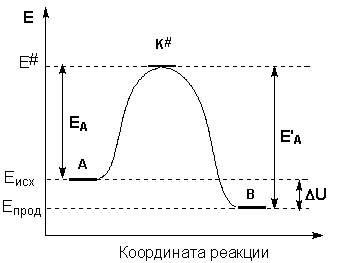
Рис. 2.5 Энергетическая диаграмма химической реакции.
Eисх – средняя энергия частиц исходных веществ,
Eпрод – средняя энергия частиц продуктов реакции
Поскольку температура есть мера средней кинетической энергии частиц, повышение температуры приводит к увеличению доли частиц, энергия которых равна или больше энергии активации, что приводит к увеличению константы скорости реакции (рис.2.6):
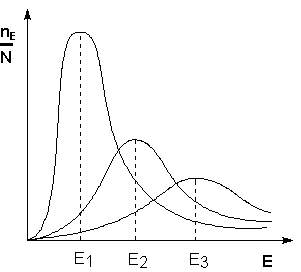
Рис. 2.6 Распределение частиц по энергии Здесь nЕ/N – доля частиц, обладающих энергией E; Ei - средняя энергия частиц при температуре Ti (T1 < T2 < T3)
Рассмотрим термодинамический вывод выражения, описывающего зависимость константы скорости реакции от температуры и величины энергии активации – уравнения Аррениуса. Согласно уравнению изобары Вант-Гоффа,
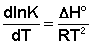 (II.31)
(II.31)
Поскольку константа равновесия есть отношение констант скоростей прямой и обратной реакции, можно переписать выражение (II.31) следующим образом:
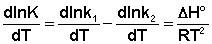 (II.32)
(II.32)
Представив изменение энтальпии реакции ΔHº в виде разности двух величин E1 и E2, получаем:
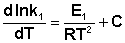 (II.33)
(II.33)
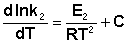 (II.34)
(II.34)
Здесь С – некоторая константа. Постулировав, что С = 0, получаем уравнение Аррениуса, где EA – энергия активации:
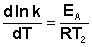 (II.35)
(II.35)
После неопределенного интегрирования выражения (II.35) получим уравнение Аррениуса в интегральной форме:
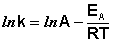 (II.36)
(II.36)
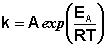 (II.37)
(II.37)
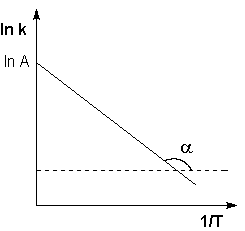
Рис. 2.7 Зависимость логарифма константы скорости химической реакции от обратной температуры.
Здесь A – постоянная интегрирования. Из уравнения (II.37) нетрудно показать физический смысл предэкспоненциального множителя A, который равен константе скорости реакции при температуре, стремящейся к бесконечности. Как видно из выражения (II.36), логарифм константы скорости линейно зависит от обратной температуры (рис.2.7); величину энергии активации EA и логарифм предэкспоненциального множителя A можно определить графически (тангенс угла наклона прямой к оси абсцисс и отрезок, отсекаемый прямой на оси ординат).
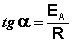 (II.38)
(II.38)
Зная энергию активации реакции и константу скорости при какой-либо температуре T1, по уравнению Аррениуса можно рассчитать величину константы скорости при любой температуре T2:
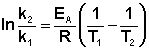 (II.39)
(II.39)
2.1.11 Кинетика двусторонних (обратимых) реакций
Химические реакции часто являются двусторонними (обратимыми), т.е. могут протекать при данных условиях в двух противоположных направлениях (понятие обратимая реакция следует отличать от термодинамического понятия обратимый процесс; двусторонняя реакция обратима в термодинамическом смысле лишь в состоянии химического равновесия). Рассмотрим элементарную двустороннюю реакцию
А + В <––> D + E
Скорость уменьшения концентрации вещества А при протекании прямой реакции определяется уравнением (II.40):
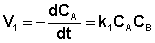 , (II.40)
, (II.40)
а скорость возрастания концентрации вещества А в результате протекания обратной реакции – уравнением (II.41):
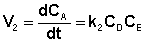 (II.41)
(II.41)
Общая скорость двусторонней реакции в любой момент времени равна разности скоростей прямой и обратной реакции:
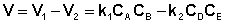 (II.42)
(II.42)
По мере протекания двусторонней реакции скорость прямой реакции уменьшается, скорость обратной реакции – увеличивается; в некоторый момент времени скорости прямой и обратной реакции становятся равными и концентрации реагентов перестают изменяться. Таким образом, в результате протекания в закрытой системе двусторонней реакции система достигает состояния химического равновесия; при этом константа равновесия будет равна отношению констант скоростей прямой и обратной реакции:
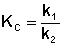 (II.43)
(II.43)
2.1.12 Кинетика гетерогенных химических реакций
Когда реакция совершается между веществами, находящимися в разных фазах гетерогенной системы, основной постулат химической кинетики становится неприменимым. В гетерогенных реакциях роль промежуточных продуктов обычно играют молекулы, связанные химическими силами с поверхностью раздела фаз (химически адсорбированные на поверхности). Во всяком гетерогенном химическом процессе можно выделить следующие стадии:
1. Диффузия реагентов к реакционной зоне, находящейся на поверхности раздела фаз.
2. Активированная адсорбция частиц реагентов на поверхности.
3. Химическое превращение адсорбированных частиц.
4. Десорбция образовавшихся продуктов реакции.
5. Диффузия продуктов реакции из реакционной зоны.
Стадии 1 и 5 называются диффузионными, стадии 2, 3 и 4 – кинетическими. Универсального выражения для скорости гетерогенных химических реакций не существует, поскольку каждая из выделенных стадий может являться лимитирующей. Как правило, при низких температурах скорость гетерогенной реакции определяют кинетические стадии (т.н. кинетическая область гетерогенного процесса; скорость реакции в этом случае сильно зависит от температуры и величины площади поверхности раздела фаз; порядок реакции при этом может быть любым). При высоких температурах скорость процесса будет определяться скоростью диффузии (диффузионная область гетерогенной реакции, характеризующаяся, как правило, первым порядком реакции и слабой зависимостью скорости процесса от температуры и площади поверхности раздела фаз).
2.2 ФОТОХИМИЧЕСКИЕ РЕАКЦИИ
Передача энергии для активации вступающих во взаимодействие молекул может осуществляться либо в форме теплоты (т. н. темновые реакции), либо в виде квантов электромагнитного излучения. Реакции, в которых активация частиц является результатом их взаимодействия с квантами электромагнитного излучения видимой области спектра, называют фотохимическими реакциями. При всех фотохимических процессах выполняется закон Гротгуса:
Химическое превращение вещества может вызвать только то излучение, которое поглощается этим веществом.
Излучение, отражённое веществом, а также прошедшее сквозь него, не вызывают никаких химических превращений. Иногда фотохимические процессы происходят под действием излучения, которое не поглощается реагирующими веществами; однако в таких случаях реакционная смесь должна содержать т.н. сенсибилизаторы. Механизм действия сенсибилизаторов заключается в том, что они поглощают свет, переходя в возбуждённое состояние, а затем при столкновении с молекулами реагентов передают им избыток своей энергии. Сенсибилизатором фотохимических реакций является, например, хлорофилл (см. ниже).
Взаимодействие света с веществом может идти по трём возможным направлениям:
1. Возбуждение частиц (переход электронов на вышележащие орбитали):
A + hν ––> A*
2. Ионизация частиц за счет отрыва электронов:
A + hν ––> A+ + e–
3. Диссоциация молекул с образованием свободных радикалов (гомолитическая) либо ионов (гетеролитическая):
AB + hν ––> A• + B•
AB + hν ––> A+ + B–
Между количеством лучистой энергии, поглощенной молекулами вещества, и количеством фотохимически прореагировавших молекул существует соотношение, выражаемое законом фотохимической эквивалентности Штарка – Эйнштейна:
Число молекул, подвергшихся первичному фотохимическому превращению, равно числу поглощенных веществом квантов электромагнитного излучения.
Поскольку фотохимическая реакция, как правило, включает в себя и т.н. вторичные процессы (например, в случае цепного механизма), для описания реакции вводится понятие квантовый выход фотохимической реакции:
Квантовый выход фотохимической реакции γ есть отношение числа частиц, претерпевших превращение, к числу поглощенных веществом квантов света.
Квантовый выход реакции может варьироваться в очень широких пределах: от 10-3 (фотохимическое разложение метилбромида) до 106 (цепная реакция водорода с хлором); в общем случае, чем более долгоживущей является активная частица, тем с большим квантовым выходом протекает фотохимическая реакция.
Важнейшими фотохимическими реакциями являются реакции фотосинтеза, протекающие в растениях с участием хлорофилла:
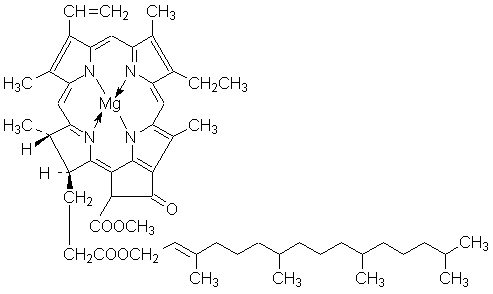
Процесс фотосинтеза составляют две стадии: световая, связанная с поглощением фотонов, и значительно более медленная темновая, представляющая собой ряд химических превращений, осуществляемых в отсутствие света. Суммарный процесс фотосинтеза заключается в окислении воды до кислорода и восстановлении диоксида углерода до углеводов:
СО2 + Н2О + hν ––> (СН2О) + О2, ΔG° = 477.0 кДж/моль
Протекание данного окислительно-восстановительного процесса (связанного с переносом электронов) возможно благодаря наличию в реакционном центре хлорофилла Сhl донора D и акцептора A электронов; перенос электронов происходит в результате фотовозбуждения молекулы хлорофилла:
DChlA + hν ––> DChl*A ––> DChl+A– ––> D+ChlA–
Возникающие в данном процессе заряженные частицы D+ и A– принимают участие в дальнейших окислительно-восстановительных реакциях темновой стадии фотосинтеза.
2.3 КАТАЛИТИЧЕСКИЕ ПРОЦЕССЫ
Скорость химической реакции при данной температуре определяется скоростью образования активированного комплекса, которая, в свою очередь, зависит от величины энергии активации. Во многих химических реакциях в структуру активированного комплекса могут входить вещества, стехиометрически не являющиеся реагентами; очевидно, что в этом случае изменяется и величина энергии активации процесса. В случае наличия нескольких переходных состояний реакция будет идти в основном по пути с наименьшим активационным барьером.
Катализ – явление изменения скорости химической реакции в присутствии веществ, состояние и количество которых после реакции остаются неизменными.
Различают положительный и отрицательный катализ (соответственно увеличение и уменьшение скорости реакции), хотя часто под термином "катализ" подразумевают только положительный катализ; отрицательный катализ называют ингибированием.
Вещество, входящее в структуру активированного комплекса, но стехиометрически не являющееся реагентом, называется катализатором. Для всех катализаторов характерны такие общие свойства, как специфичность и селективность действия.
Специфичность катализатора заключается в его способности ускорять только одну реакцию или группу однотипных реакций и не влиять на скорость других реакций. Так, например, многие переходные металлы (платина, медь, никель, железо и т.д.) являются катализаторами для процессов гидрирования; оксид алюминия катализирует реакции гидратации и т.д.
Селективность катализатора – способность ускорять одну из возможных при данных условиях параллельных реакций. Благодаря этому можно, применяя различные катализаторы, из одних и тех же исходных веществ получать различные продукты:
| [Cu]:СО + Н2 ––> СН3ОН | [Al2О3]: С2Н5ОН ––> С2Н4 + Н2О |
| [Ni]: СО + Н2 ––> СН4 + Н2О | [Cu]: С2Н5ОН ––> СН3СНО + Н2 |
Причиной увеличения скорости реакции при положительном катализе является уменьшение энергии активации при протекании реакции через активированный комплекс с участием катализатора (рис. 2.8).
Поскольку, согласно уравнению Аррениуса, константа скорости химической реакции находится в экспоненциальной зависимости от величины энергии активации, уменьшение последней вызывает значительное увеличение константы скорости. Действительно, если предположить, что предэкспоненциальные множители в уравнении Аррениуса (II.32) для каталитической и некаталитической реакций близки, то для отношения констант скорости можно записать:
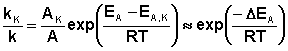 (II.44)
(II.44)
Если ΔEA = –50 кДж/моль, то отношение констант скоростей составит 2,7·106 раз (действительно, на практике такое уменьшение EA увеличивает скорость реакции приблизительно в 105 раз).
Необходимо отметить, что наличие катализатора не влияет на величину изменения термодинамического потенциала в результате процесса и, следовательно, никакой катализатор не может сделать возможным самопроизвольное протекание термодинамически невозможного процесса (процесса, ΔG (ΔF) которого больше нуля). Катализатор не изменяет величину константы равновесия для обратимых реакций; влияние катализатора в этом случае заключается только в ускорении достижения равновесного состояния.
В зависимости от фазового состояния реагентов и катализатора различают гомогенный и гетерогенный катализ.
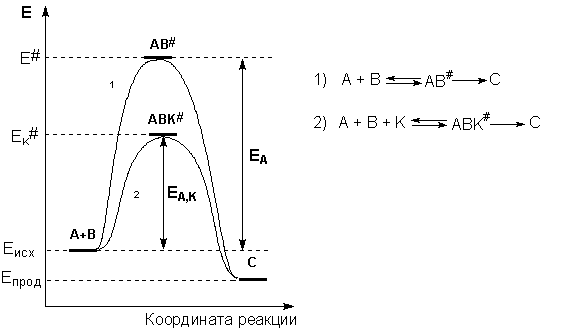
Рис. 2.8 Энергетическая диаграмма химической реакции без катализатора (1) и в присутствии катализатора (2).
2.3.1 Гомогенный катализ.
Гомогенный катализ – каталитические реакции, в которых реагенты и катализатор находятся в одной фазе. В случае гомогенно-каталитических процессов катализатор образует с реагентами промежуточные реакционноспособные продукты. Рассмотрим некоторую реакцию
А + В ––> С
В присутствии катализатора осуществляются две быстро протекающие стадии, в результате которых образуются частицы промежуточного соединения АК и затем (через активированный комплекс АВК#) конечный продукт реакции с регенерацией катализатора:
А + К ––> АК
АК + В ––> С + К
Примером такого процесса может служить реакция разложения ацетальдегида, энергия активации которой EA = 190 кДж/моль:
СН3СНО ––> СН4 + СО
В присутствии паров йода этот процесс протекает в две стадии:
СН3СНО + I2 ––> СН3I + НI + СО
СН3I + НI ––> СН4 + I2
Уменьшение энергии активации этой реакции в присутствии катализатора составляет 54 кДж/моль; константа скорости реакции при этом увеличивается приблизительно в 105 раз. Наиболее распространенным типом гомогенного катализа является кислотный катализ, при котором в роли катализатора выступают ионы водорода Н+.
2.3.2Автокатализ.
Автокатализ – процесс каталитического ускорения химической реакции одним из её продуктов. В качестве примера можно привести катализируемую ионами водорода реакцию гидролиза сложных эфиров. Образующаяся при гидролизе кислота диссоциирует с образованием протонов, которые ускоряют реакцию гидролиза. Особенность автокаталитической реакции состоит в том, что данная реакция протекает с постоянным возрастанием концентрации катализатора. Поэтому в начальный период реакции скорость её возрастает, а на последующих стадиях в результате убыли концентрации реагентов скорость начинает уменьшаться; кинетическая кривая продукта автокаталитической реакции имеет характерный S-образный вид (рис. 2.9).
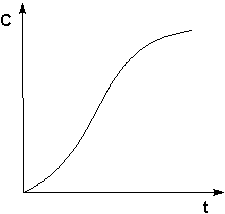
Рис. 2.9 Кинетическая кривая продукта автокаталитической реакции
2.3.3 Гетерогенный катализ.
Гетерогенный катализ – каталитические реакции, идущие на поверхности раздела фаз, образуемых катализатором и реагирующими веществами. Механизм гетерогенно-каталитических процессов значительно более сложен, чем в случае гомогенного катализа. В каждой гетерогенно-каталитической реакции можно выделить как минимум шесть стадий:
1. Диффузия исходных веществ к поверхности катализатора.
2. Адсорбция исходных веществ на поверхности с образованием некоторого промежуточного соединения:
А + В + К ––> АВК
3. Активация адсорбированного состояния (необходимая для этого энергия есть истинная энергия активации процесса):
АВК ––> АВК#
4. Распад активированного комплекса с образованием адсорбированных продуктов реакции:
АВК# ––> СDК
5. Десорбция продуктов реакции с поверхности катализатора.
СDК ––> С + D + К
6. Диффузия продуктов реакции от поверхности катализатора.
Специфической особенностью гетерокаталитических процессов является способность катализатора к промотированию и отравлению.
Промотирование – увеличение активности катализатора в присутствии веществ, которые сами не являются катализаторами данного процесса (промоторов). Например, для катализируемой металлическим никелем реакции
СО + Н2 ––> СН4 + Н2О
введение в никелевый катализатор небольшой примеси церия приводит к резкому возрастанию активности катализатора.
Отравление – резкое снижение активности катализатора в присутствии некоторых веществ (т. н. каталитических ядов). Например, для реакции синтеза аммиака (катализатор – губчатое железо), присутствие в реакционной смеси соединений кислорода или серы вызывает резкое снижение активности железного катализатора; в то же время способность катализатора адсорбировать исходные вещества снижается очень незначительно.
Для объяснения этих особенностей гетерогенно-каталитических процессов Г. Тэйлором было высказано следующее предположение: каталитически активной является не вся поверхность катализатора, а лишь некоторые её участки – т.н. активные центры, которыми могут являться различные дефекты кристаллической структуры катализатора (например, выступы либо впадины на поверхности катализатора). В настоящее время нет единой теории гетерогенного катализа. Для металлических катализаторов была разработана теория мультиплетов. Основные положения мультиплетной теории состоят в следующем:
1. Активный центр катализатора представляет собой совокупность определенного числа адсорбционных центров, расположенных на поверхности катализатора в геометрическом соответствии со строением молекулы, претерпевающей превращение.
2. При адсорбции реагирующих молекул на активном центре образуется мультиплетный комплекс, в результате чего происходит перераспределение связей, приводящее к образованию продуктов реакции.
Теорию мультиплетов называют иногда теорией геометрического подобия активного центра и реагирующих молекул. Для различных реакций число адсорбционных центров (каждый из которых отождествляется с атомом металла) в активном центре различно – 2, 3, 4 и т.д. Подобные активные центры называются соответственно дублет, триплет, квадруплет и т.д. (в общем случае мультиплет, чему и обязана теория своим названием).
Например, согласно теории мультиплетов, дегидрирование предельных одноатомных спиртов происходит на дублете, а дегидрирование циклогексана – на секстете (рис. 2.10 – 2.11); теория мультиплетов позволила связать каталитическую активность металлов с величиной их атомного радиуса.
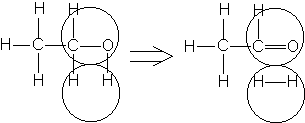 Рис. 2.10 Дегидрирование спиртов на дублете
Рис. 2.10 Дегидрирование спиртов на дублете
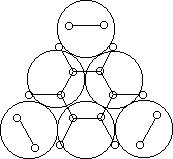
Рис. 2.11 Дегидрирование циклогексана на секстете
2.3.4 Ферментативный катализ.
Ферментативный катализ – каталитические реакции, протекающие с участием ферментов – биологических катализаторов белковой природы. Ферментативный катализ имеет две характерные особенности:
1. Высокая активность, на несколько порядков превышающая активность неорганических катализаторов, что объясняется очень значительным снижением энергии активации процесса ферментами. Так, константа скорости реакции разложения перекиси водорода, катализируемой ионами Fе2+, составляет 56 с-1; константа скорости этой же реакции, катализируемой ферментом каталазой, равна 3.5·107, т.е. реакция в присутствии фермента протекает в миллион раз быстрее (энергии активации процессов составляют соответственно 42 и 7.1 кДж/моль). Константы скорости гидролиза мочевины в присутствии кислоты и уреазы различаются на тринадцать порядков, составляя 7.4·10-7 и 5·106 с-1 (величина энергии активации составляет соответственно 103 и 28 кДж/моль).
2. Высокая специфичность. Например, амилаза катализирует процесс расщепления крахмала, представляющего собой цепь одинаковых глюкозных звеньев, но не катализирует гидролиз сахарозы, молекула которой составлена из глюкозного и фруктозного фрагментов.
Согласно общепринятым представлениям о механизме ферментативного катализа, субстрат S и фермент F находятся в равновесии с очень быстро образующимся фермент-субстратным комплексом FS, который сравнительно медленно распадается на продукт реакции P с выделением свободного фермента; т.о., стадия распада фермент-субстратного комплекса на продукты реакции является скоростьопределяющей (лимитирующей).
F + S <––> FS ––> F + P
Исследование зависимости скорости ферментативной реакции от концентрации субстрата при неизменной концентрации фермента показали, что с увеличением концентрации субстрата скорость реакции сначала увеличивается, а затем перестает изменяться (рис. 2.12) и зависимость скорости реакции от концентрации субстрата описывается следующим уравнением:
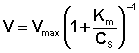 (II.45)
(II.45)
Здесь Кm – константа Михаэлиса, численно равная концентрации субстрата при V = ½Vmax. Константа Михаэлиса служит мерой сродства между субстратом и ферментом: чем меньше Кm, тем больше их способность к образованию фермент-субстратного комплекса.
Характерной особенностью действия ферментов является также высокая чувствительность активности ферментов к внешним условиям – рН среды и температуре. Ферменты активны лишь в достаточно узком интервале рН и температуры, причем для ферментов характерно наличие в этом интервале максимума активности при некотором оптимальном значении рН или температуры; по обе стороны от этого значения активность ферментов быстро снижается.
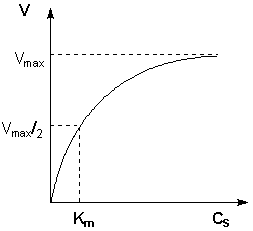
Рис. 2.12 Зависимость скорости ферментативной реакции от концентрации субстрата.
|
|
|
|
|
Дата добавления: 2014-12-07; Просмотров: 1188; Нарушение авторских прав?; Мы поможем в написании вашей работы!