КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Синдром ретикулярной дисгенезии
|
|
|
|
Существует множество отдельных нозологических единиц иммунодефицитов.
Примеры иммунодефицитов
Некоторые иммунодефициты представлены на рис. 16–8.
Ы ВЁРСТКА Вставить файл «ПФ Рис 16 08 Виды ИДС по преимущественному поражению клеток иммунной системы»
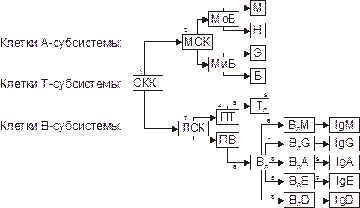
Рис. 16–8. Иммунодефициты, обусловленных блокадой созревания клеток иммунной системы (цифрами обозначены наиболее частые места блокады). Условные обозначения:1 — ретикулярная дисгенезия; 2 — моноцитопения, фагоцитарная недостаточность (синдром Шедьяка‑Хигаси); 3 — агаммаглобулинемия швейцарского типа; 4 — синдром ДиДжорджи; 5 — синдром Вискотта‑Олдрича; 6 — болезнь Брутона (агаммаглобулинемия); 7 — селективный дефицит IgG; 8 — селективный дефицит IgA, IgE, IgD; 9 — синдром Луи‑Бар. СКК — стволовая кроветворная клетка; МСК — миелостволовая клетка; ЛСК — лимфостволовая клетка; МоБ — монобласт; МиБ — миелобласт; М — моноцит; Н — нейтрофил; Э — эозинофил; Б — базофил; ПТ — клетка‑предшественница T-лимфоцитов; Тл — T-лимфоцит; ПВ — клетка‑предшественница B-лимфоцитов; ВЛ — B-лимфоцит; ВЛМ, BЛG, ВЛА, ВЛЕ, ВЛD — соответственно B-лимфоцит, продуцирующий IgМ, IgG, IgА, IgЕ, IgD.
При врождённой алейкии (ретикулярный дисгенез, *267500, ) врождённый агранулоцитоз и лейкопения приводят к развитию тяжёлого иммунодефицита, часто сочетающегося с гипоплазией вилочковой железы. Синдром ретикулярной дисгенезии (на рис. 16–8 помечен цифрой 1) характеризуется значительным уменьшением в костном мозге количества стволовых кроветворных клеток, блоком созревания из них миело‑, лимфо‑ и моноцитов с развитием комбинированного дефицита А‑, В‑ и T-клеток, а также нейтрофилов. Пациенты с этим синдромом, как правило, погибают вскоре после рождения от различных инфекций (нередко — от сепсиса) или злокачественных опухолей.
Синдром Шедьяка‑Хигаси
При аномалии Шедьяка‑Штайнбринка ‑ Хигаси (, *214450, *214500) происходит блокада пролиферации миелостволовой клетки (на рис. 16–8 помечена цифрой 2), что приводит к многочисленным последствиям: дефектам фагоцитоза, гипогаммаглобулинемии, нейтропении, тромбоцитопении. Характерны низкая активность миелопероксидазы, торможение хемотаксиса, патологические изменения гранул и ядер всех типов лейкоцитов, дефекты гранул с положительной пероксидазной реакцией, цитоплазматические включения, тельца Дёле, светлая радужная оболочка, альбинизм, возможна гиперпигментация кожи, гепатоспленомегалия, лимфоаденопатия, анемия, изменения в костях, лёгких, сердце, а также психомоторные дефекты и выраженная предрасположенность к инфекциям.
Тяжёлый комбинированный иммунодефицит
В классическом варианте отсутствует как гуморальный (нет АТ), так и клеточный иммунитет (нет Т-клеток и естественных киллеров — NK-клеток); алимфоплазия или лимфопения (относится как к B-лимфоцитами, так и к T-лимфоцитам). Характерна безусловная восприимчивость к бактериальным, грибковым, протозойным, вирусным инфекциям; введение живых вакцин должно быть исключено; смерть наступает к концу первого года жизни (если не проведена трансплантация костного мозга). Примерно у 70% больных В-лимфоциты присутствуют (в том числепри мутациях генов ИЛ, недостаточности аденозиндезаминазы, синдроме голых лимфоцитов). Типы:
• Недостаточность аденозиндезаминазы (КФ 3.5.4.4, три изоформы, дефектные варианты — *102700, 20q12–q13.11, дефект гена ADA, известно не менее 30 аллелей, , ) — причина 50% случаев тяжёлого комбинированного иммунодефицита. Проявления: B- и T-клеточный иммунодефицит, CD4+-лимфопения, тромбоцитопеническая пурпура, гепатоспленомегалия, повторяющиеся бактериальные, вирусные, грибковые инфекции (преимущественно бронхолёгочные), часты различные дисплазии костного скелета.
• Агаммаглобулинемия швейцарского типа (см. статью «Агаммаглобулинемии» в приложении «Справочник терминов»).
• Недостаточность пуриннуклеозид фосфорилазы (см. статью «Недостаточность ферментов» в приложении «Справочник терминов»).
• Дефицит транскобаламина II (*275350, 22q12–q13, дефекты генов TCN2, TC2, ), транспортного белка витамина В12. Проявления: тяжёлая мегалобластная анемия, агранулоцитоз, тромбоцитопения, геморрагический диатез, тяжёлая диарея, язвенный стоматит, повторные инфекции, агаммаглобулинемия.
• Синдром голых лимфоцитов (#209920, 600005, 600006, 601863, 601861, в том числе дефекты генов MHC2TA, RFX5, RFXAP, C2TA, все ). Термин применяют по отношению к тяжёлому комбинированному иммунодефициту с отсутствием экспрессии ряда генов MHC класса II (отсутствуют Аг HLA на поверхности лимфоидных клеток). Проявления: упорная диарея, синдром мальабсорбции, кандидоз, бактериальные инфекции, интерстициальная пневмония. Лабораторно: пангипогаммаглобулинемия, отсутствуют стимулируемая Аг пролиферация лимфоцитов и клеточноопосредованная цитотоксичность.
• Сцепленная с хромосомой X форма (#312863, мутации -цепи рецептора ИЛ2, рецессивное). Слабое развитие лимфоидной ткани, множественного генеза инфекции; нормальное содержание Ig, В- и NK-клеток, уменьшено число CD4+и CD8+T-лимфоцитов, слабый их пролиферативный ответ на Аг и митогены.
|
|
|
|
|
Дата добавления: 2015-06-04; Просмотров: 1345; Нарушение авторских прав?; Мы поможем в написании вашей работы!