КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Изменение энтропии в химических реакциях
|
|
|
|
Изменение энтальпии в химических реакциях.
1. Понятия и определения химической термодинамики
Химическая термодинамика – наука о зависимости направления и пределов превращений веществ от условий, в которых эти вещества находятся. Термин предложил в 1851 г. англ. Уильям Томсон (лорд Кельвин с 1892) (1824-1907), когда сформулировал второе начало. Рудольф Юлиус Эмануэль Клаузиус (1822-1888) – нем., называл новую науку "механической теорией тепла". В отличие от других разделов физической химии (строение вещества и химическая кинетика), химическую термодинамику можно применять, ничего не зная о молекулярном строении вещества. Такое описание требует значительно меньше исходных данных.
Пример: энтальпию образования глюкозы нельзя определить прямым экспериментом:
6 C + 6 H2 + 3 O2 = C6H12O6 (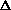 Hх -?)
Hх -?)
такая реакция невозможна
6 CO2 + 6 H2O = C6H12O6 + 6 O2 (Hу -?)
реакция идет в зеленых листьях, но вместе с другими процессами.
Пользуясь законом Гесса, достаточно скомбинировать три уравнения сжигания:
1) C + O2 = CO2
H1 = -394 кДж
2) H2 + 1/2 O2 = H2O(пар)
H2 = -242 кДж
3) C6H12O6 + 6 O2 = 6 CO2 + 6 H2O
H3 = -2816 кДж
Складываем уравнения, "разворачивая" третье, тогда
Hх = 6 H1 + 6 H2 - H3 = 6(-394) + 6(-242) -(-2816) =
= -1000 кДж/моль
При решении не использованы никакие данные по строению глюкозы; не рассматривался также механизм ее горения.
Конкретный объект термодинамического исследования называют термодинамической системой или просто системой, выделенной из окружающего мира реально существующими или воображаемыми поверхностями. Системой может быть газ в сосуде, раствор реагентов в колбе, кристалл вещества или даже мысленно выделенная часть этих объектов.
Для того чтобы систему можно было описать термодинамически, она должна состоять из большого числа частиц – соответствовать законам статистики. Если в системе есть реальные поверхности раздела, отделяющие друг от друга части системы, различающиеся по свойствам, то система называется гетерогенной (насыщенный раствор с осадком), если таких поверхностей нет, система называется гомогенной (истинный раствор). Гетерогенные системы содержат не менее двух фаз. Фаза – совокупность всех гомогенных частей системы, одинаковых по составу и по всем физическим и химическим свойствам (не зависящим от количества вещества) и отграниченных от других частей системы поверхностью раздела. Внутри одной фазы свойства могут изменяться непрерывно, но на поверхности раздела между фазами свойства меняются скачком. Пример двухфазной системы – поверхность реки в ледоход. Компонентами называют вещества, минимально необходимые для составления данной системы (минимум один). Число компонентов в системе равно числу веществ в ней присутствующих, минус число связывающих эти вещества независимых уравнений. По уровням взаимодействия с окружающей средой термодинамические системы принято делить на:
– открытые – обмениваются с окружающей средой веществом и энергией (например, живые объекты);
– закрытые – обмениваются только энергией (например, реакция в закрытой колбе или колбе с обратным холодильником), наиболее частый объект химической термодинамики;
– изолированные – не обмениваются ни веществом, ни энергией и сохраняют постоянный объем (приближение – реакция в термостате).
Свойства системы разделяют на экстенсивные (суммирующиеся) – например, общий объем, масса, и интенсивные (выравнивающиеся) – давление, температура, концентрация и т.п. Совокупность свойств системы определяет ее состояние. Многие свойства взаимосвязаны, поэтому для гомогенной однокомпонентной системы с известным количеством вещества n достаточно выбрать для характеристики состояния два из трех свойств: температуру T, давление p и объем V. Связывающее свойства уравнение называют уравнением состояния, для идеального газа это: pV = nRT. Наиболее важны для расчетов – функции состояния – такие термодинамические функции, значения которых зависят только от состояния системы и не зависят от пути перехода между состояниями. Процесс в термодинамике – это не развитие события во времени, а последовательность равновесных состояний системы, ведущих от начального набора термодинамических переменных к конечному. Термодинамика позволяет полностью решить поставленную задачу, если исследуемый процесс в целом описывается совокупностью равновесных стадий. Например, работа реактивного двигателя – это последовательность почти равновесных процессов в каждом малом сечении двигателя (быстрые реакции быстро устанавливают равновесие). В расчетах используют численные данные (табличные) о термодинамических свойствах веществ. Даже небольшие наборы таких данных позволяют рассчитывать множество различных процессов. Для расчета равновесного состава не требуется записывать уравнения возможных химических реакций, достаточно учесть все вещества, которые могут в принципе составлять равновесную смесь. Таким образом, химическая термодинамика не дает чисто расчетного (неэмпирического) ответа на вопрос "почему?" и тем более "как?"; она решает задачи по принципу "если..., то...".
2. Изменение энтальпии в химических реакциях
В большинстве курсов химической термодинамики рассматривается три закона. Однако для строгого определения термического равновесия в 1931 г. англ. Р.Фаулер сформулировал закон, который называют нулевым: Две системы, находящиеся в термическом равновесии с третьей системой, состоят в термическом равновесии друг с другом.
Первый закон термодинамики – одна из форм закона сохранения энергии.
Его формулировки:
Энергия не создается и не уничтожается.
Вечный двигатель (perpetuum mobile) первого рода невозможен.
В любой изолированной системе общее количество энергии постоянно.
Энергетический эффект химической реакции может проявляться как чисто тепловой, связанный с изменением внутренней энергии системы, например реакция нейтрализации в разбавленном растворе: H++OH-=H2O+57кДж
Для этого случая можно записать, что весь тепловой эффект Q при постоянном объеме равен изменению внутренней энергии
U: Qv = U
Однако если смешать в пробирке водные растворы карбоната натрия и соляной кислоты и быстро закрыть пробирку пробкой, то через некоторое время система совершит механическую работу, "выстрелив" пробкой. При этом температура растворов после реакции практически не изменяется. Работа совершается, когда повышенное давление в закрытой пробирке уравнивается с атмосферным после вылетания пробки. Таким образом, можно описать работу, как работу расширения газа, совершенную при постоянном давлении (изобарный процесс): p V
В общем случае, работа, совершаемая химической реакцией при постоянном давлении, состоит из изменения внутренней энергии и работы расширения: Qp = U + p V
Для большинства химических реакций, проводимых в открытых сосудах, удобно использовать функцию состояния, приращение которой равно теплоте, полученной системой в изобарном процессе. Эта функция называется энтальпия (от греч. "энтальпо" – нагреваю):
Qp = H = U + p V
Другое определение: разность энтальпий в двух состояниях системы равна тепловому эффекту изобарного процесса.
Существуют обширные таблицы, содержащие данные по стандартным энтальпиям образования веществ Ho298. Индексы означают, что для химических соединений приведены энтальпии образования 1 моль их из простых веществ, взятых в наиболее устойчивой модификации (кроме белого фосфора – не самой устойчивой, а самой воспроизводимой формы фосфора) при 1 атм (1,01325.105 Па или 760 мм.рт.ст) и 298,15 К (25оС). Если речь идет об ионах в растворе, то стандартной является концентрация 1 М (1 моль/л). В принципе, можно попытаться вычислить абсолютные значения энтальпий для химии (тепловой эффект образования 1 моль соединения из бесконечно удаленных атомов, взятых при 0оК) или для физики (исходя из элементарных частиц, взятых при 0оК), но для реальных расчетов общепринятый произвольный уровень отсчета вполне удобен. Знак энтальпии определяется "с точки зрения" самой системы: при выделении теплоты изменение энтальпии отрицательно, при поглощении теплоты изменение энтальпии положительно.
Вернемся теперь к реакции раствора соды с раствором соляной кислоты:
Na2CO3 + 2 HCl = 2 NaCl + H2O + CO2
Для такой записи мы скорее всего не найдем нужных табличных данных – есть значения Ho298 для твердых солей и газообразного хлороводорода, а наша реакция происходила при сливании двух растворов. Чтобы произвести правильный расчет, нужно определить, что на самом деле реагирует:
CO32- + 2 H+ = H2O(ж) + CO2
Ho298,кДж/моль -677 0 -286 -394
По закону Гесса получаем для реакции Ho298 = -3 кДж.
3. Изменение энтропии в химических реакциях
Вполне очевидно, что реакции с суммарным уменьшением энтальпии (экзотермические) могут идти самопроизвольно, как катящийся с горы камень. Однако хорошо известно, что самопроизвольно идут также некоторые реакции, сопровождающиеся увеличением энтальпии и охлаждением реактора (эндотермические).
Для характеристики эндотермических процессов и определения условий их самопроизвольного осуществления была введена новая функция состояния – энтропия (от греч. "эн" – "в", "внутрь" и "тропе" – "поворот", "превращение"). Изменение энтропии равно (по определению) минимальной теплоте, подводимой к системе в обратимом (все промежуточные состояния равновесны) изотермическом процессе, деленной на абсолютную температуру процесса:
S = Qмин./T
На данном этапе изучения термодинамики следует принять как постулат, что существует некоторое экстенсивное свойство системы S, называемое энтропией, изменение которого так связано с процессами в системе:
В самопроизвольном процессе S > Qмин./T
В равновесном процессе S = Qмин./T
В несамопроизвольном процессе S < Qмин./T
Для изолированной системы, где dQ = 0, получим:
В самопроизвольном процессе S > 0
В равновесном процессе S = 0
В несамопроизвольном процессе S < 0
В общем случае энтропия изолированной системы или увеличивается, или остается постоянной.
Понятие энтропии возникло из полученных ранее формулировок второго закона (начала) термодинамики. Энтропия – свойство системы в целом, а не отдельной частицы. Второе начало по У.Томсону (1851): "в природе невозможен процесс, единственным результатом которого была бы механическая работа, совершенная за счет охлаждения теплового резервуара". По Р.Клаузиусу (1850): " теплота сама по себе не может перейти от более холодного тела к более теплому " или: "невозможно сконструировать машину, которая, действуя посредством кругового процесса, будет только переносить теплоту с более холодного тела на более теплое".
Самая ранняя формулировка второго начала термодинамики появилась раньше первого начала, на основании работы фр. С.Карно (1824) и ее математической интерпретации фр. Э.Клапейроном (1834) как КПД идеальной тепловой машины:
КПД = (T1 - T2)/T1
Карно и Клапейрон сформулировали закон сохранения теплорода – невесомой неуничтожимой жидкости, содержание которой определяет температуру тела. Теория теплорода господствовала в термодинамике до середины XIX века, при этом законы и соотношения, выведенные на основе представлений о теплороде, оказались действительными и в рамках молекулярно-кинетической теории теплоты. В 1911 г. Макс Планк предложил следующий постулат: энтропия правильно сформированного кристалла чистого вещества при абсолютном нуле равна нулю. Этот постулат может быть объяснен статистической термодинамикой, согласно которой энтропия есть мера беспорядочности системы на микроуровне: S = kblnW " уравнение Больцмана ", выведено М.Планком в 1900 г. W – число различных состояний системы, доступное ей при данных условиях, или термодинамическая вероятность макросостояния системы. kb = R/NA = 1,38.10-16 эрг/град – постоянная Больцмана. В 1872 г. Л.Больцман предложил статистическую формулировку второго закона термодинамики: изолированная система эволюционирует преимущественно в направлении большей термодинамическоой вероятности.
Следует всегда помнить, что второй закон термодинамики не является абсолютным; он теряет смысл для систем, содержащих малое число частиц, и для систем космического масштаба. Второй закон, особенно в статистической формулировке, неприменим к живым объектам, которые представляют собой открытые системы и постоянно уменьшают энтропию, создавая идеально упорядоченные молекулы, например, за счет энергии солнечного света. Введение энтропии дало возможность установить критерии, позволяющие определить направление и глубину протекания любого химического процесса (для большого числа частиц в равновесии). Макроскопические системы достигают равновесия, когда изменение энергии компенсируется энтропийной составляющей:
При постоянном объеме и температуре:
Uv = T Sv или (U-TS) F = 0 энергия Гельмгольца или изохорно-изо-термический потенциал
При постоянном давлении и температуре:
Hp = T Sp или (H-TS) G = 0 энергия Гиббса или свободная энергия Гиббса или изобарно-изотермический потенциал
Изменение энергии Гиббса как критерий возможности химической реакции
Для данной температуры G = H - T S
При G < 0 реакция возможна;
при G > 0 реакция невозможна;
при G = 0 система находится в равновесии.
Возможность самопроизвольной реакции в изолированной системе определяется сочетанием знаков энергетического (энтальпийного) и энтропийного факторов:
Знак H + - - +
Знак S - + - +
Возможность
самопроизвольной реакции Нет Да Зависит
от соотношения
H и T S
Имеются обширные табличные данные по стандартным значениям G0 и S0 (для энтропии по третьему закону есть нулевой уровень отсчета и соответственно абсолютные значения), позволяющие вычислить G0 реакции.
В случае, если температура отличается от 298 К и концентрации реагентов – от 1 М, для процесса в общем виде:
aA + bB cC + dD
G = G0 + RT ln([C]c[D]d/[A]a[B]b)
В положении равновесия G = 0 и G0 = -RTlnKр, где
Kр = [C]cравн[D]dравн/[A]aравн[B]bравн константа равновесия
Kр = exp(-G0/RT)
Пользуясь приведенными формулами, можно определить температуру, начиная с которой эндотермическая реакция, при которой возрастает энтропия, становится легко осуществимой. Температура определяется из условия: G0 = H0 - T S0 = 0; T = H0/ S0
Следует учесть, что для точных расчетов при температурах, заметно отличающихся от 298 К, необходимо использовать для всех термодинамических функций их зависимости от температуры. Например, для реакции: 2 CH4 = C2H4 + 2 H2
В первом приближении H0 = 48,3 ккал/моль C2H4; во втором приближении HT = 46370 + 6,47 T кал/моль
|
|
|
|
|
Дата добавления: 2014-01-07; Просмотров: 3191; Нарушение авторских прав?; Мы поможем в написании вашей работы!